HEMATOPOYESIS
|

Fase megaloblástica: Los primeros
indicios de hematopoyesis se encuentran ya en la tercera semana de la gestación
en la mesénquima del saco vitelino.
Fase hepatoesplénica: a partir de la
sexta semana de la gestación ocurre en el hígado y poco tiempo después también
en el bazo.
Fase medular: A partir del quinto
mes de embarazo comienza la hematopoyesis en la médula ósea y con ella también
se inicia la formación de los leucocitos. (Manascero 2,008)
Eritropoyesis
Es el término usado para designar la actividad proliferativa
eritroide de la médula ósea, su factor
estimulante es la eritropoyetina, la cual debe de estar en los niveles altos
para proteger a los eritroblastos contra la apoptosis; todas las células de la
hematopoyesis tienen como célula de origen la célula madre hematopoyética, y a
partir de ella deriva hacia el linaje mieloide la célula madre mieloide que da
origen a la primera célula comprometida exclusivamente a la producción de los
eritrocitos la Célula Progenitora
Eritroide, que da origen a las siguientes células:
Proeritroblasto:
o Tamaño:
Grande de 20 25 µ
o Forma: Grande
redondeada.
o Descripción del
núcleo y citoplasma: El núcleo redondo
central de gran talla que ocupa mayor parte de la célula.La cromatina muestra
una estructura finamente reticulada, y posee un o dos núcleos mal limitados. El
citoplasma es intensamente basófilo debido a su gran riqueza en polirribosomas,
y queda reducida a una delgada franja perinuclear en lo que se aprecia un zona
más clara, de forma semilunar, que corresponde al centrosoma de la célula.
o Relación
núcleo/citoplasma: Alta, el núcleo
redondo central de gran talla que ocupa mayor parte de la célula, (5:1 a 8:1)
o Organelas y/o
inclusiones evidentes en la misma: En
ocasiones presenta unas protuberancias citoplasmáticas a modo de casquetes
bastante característicos de este estadio madurativo. No posee nucléolos.
o Sitio del cuerpo
donde se pueden encontrar normalmente:
Médula ósea.
 |
| Recuperado de: http://www.telmeds.org/atlas/
hematologia/serie-roja/ontogenia
-de-serie-eritroide/proeritroblasto/
|
Normoblasto
basófilo:
o Tamaño: 16
a 18 µ
o Forma: Célula
redondeada de menor tamaño que el proeritroblasto.
o Descripción del
núcleo y citoplasma: Posee un núcleo
central, pero su cromatina es algo madura, ya que se observan algunas
condensaciones cromatínicas que ocultan la visibilidad del núcleolo. Pueden
notarse unas pocas masas de cromatina aglutinadas a lo largo del borde de la
membrana nuclear. El citoplasma todavía tiene un color basófilo intenso.
o Relación
núcleo/citoplasma: Menor que la del
pronormoblasto, pero mayor que la del mieloblasto.
o Organelas y/o
inclusiones evidentes en la misma La cromatina en el núcleo adquiere un patrón
más tosco, uno o ningún nucleolo.
o Sitio del cuerpo
donde se pueden encontrar normalmente: Médula
ósea. (Manascero 2,008)
.jpg) |
| Recuperado de: http://chiquitbb.blogspot.com/2010/09/maduracion-de-los-eritrocitos.html |
Normoblasto
prolicromatofilo:
o Tamaño: 8
a 12 µ.
o Forma: Redondeada
o ligeramente oval.
o Descripción del
núcleo y citoplasma: El núcleo es
redondo y la cromatina está fuertemente condensada, habitualmente extrínseco,
membrana nuclear gruesa. El citoplasma en que se ha iniciado poco a poco la
síntesis hemoglobinica, pierde basofilia por la disminución de ribosomas y
adquiere una tonalidad gris rosada acidófila, conferida por la hemoglobina. Es
la última célula de esta línea con capacidad mitótica.
o Relación
núcleo/citoplasma: Menor que el de sus
precursores. (1:1 y 4:1)
o Organelas y/o inclusiones
evidentes en la misma: Se caracteriza por
la aparición inicial de la hemoglobina
con disposición perinuclear. La cromatina esta netamente definida, al
contrario en el linfocito, con el que se puede llegar a confundir, si no se
descubre la existencia de hemoglobina.
o Sitio del cuerpo
donde se pueden encontrar normalmente: Médula
ósea.
 |
| Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-roja/ontogenia-de-serie-eritroide/eritroblasto-policromatico/ |
Normoblasto
ortoromatico:
o Tamaño: 7
a 10 µ.
o Forma: Redondeada
u ovalada.
o Descripción del
núcleo y citoplasma: El núcleo está en
posición excéntrica o ligeramente excluido, es intensamente picnótico
(fragmentado y sin estructura), lo que le da un aspecto de cromatina homogénea
y muy condensada. El citoplasma es muy acidófilo dado el aumento en el
contenido hemoglobínico, hasta adquirir la tonalidad propia del hematíe maduro.
No posee capacidad mitótica, pero puede sintetizar hemoglobina, una vez
finalizada su maduración, el núcleo es exocitado, para luego ser fagocitado por
las células del sistema fagocítico monuclear de la médula ósea.
o Relación
núcleo/citoplasma: Menor relación
núcleo citoplasma. (1:4 a 1:2)
o Organelas y/o
inclusiones evidentes en la misma: Son
reconocibles pocas estructuras; ya que no es posible distinguir la
paracromatina. No posee nucléolos.
o Sitio del cuerpo
donde se pueden encontrar normalmente: En las extensiones de médula ósea es difícil
encontrar normoblastos ortocromáticos con citoplasma totalmente anaranjado. (Manascero 2,008)
 |
| Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-roja/ontogenia-de-serie-eritroide/eritroblasto-ortocromatico/ |
Reticulocito:
o Tamaño: 8
µ a 9 µ, algo superior al hematíe regular maduro.
o Forma:
Redonda ovalada,
o Descripción del
núcleo y citoplasma: Son anucleados que
todavía poseen cierta características de síntesis de ARN, proteínas y
hemoglobinas, agracias a que se mantienen algunas mitocondrias, ribosomas, y
restos de reticuloendoplasma. Conserva cierto grado de basofilia, a medida que
el reticulocito madura, va perdiendo el retículo granulofilamentoos hasta
transformarse en hematíe maduro, desprovisto de éste
o Relación
núcleo/citoplasma: No existe núcleo,
nula.
o Organelas y/o
inclusiones evidentes en la misma: algunas
mitocondrias, ribosomas, aparato de Golgi presente durante la etapa de síntesis
de hemoglobina, los ribosomas ricos en RNA presentes en los reticulocitos se
tiñen de azul con el azul de cresil brillante y se denominana sustancia
reticulofilamentosa; tiene receptores de transferrina en su membrana de tal
forma que el hierro puede entrar en la célula, la transferrina hace a los
reticulocitos más pegajosos y difiiculta su paso desde la médula hacia la
sangre periférica.
o Sitio del cuerpo
donde se pueden encontrar normalmente: Después
de 2 a 2.5 días en la médula ósea, el Reticulocito es liberado a sus senos
vasculares. De aquí logra llegar a la sangre perica allí continúa con su
maduración durante un día más. El recuento del número de reticulocitos en
sangre periférica es más un dato muy útil para establecer el índice de
efectividad global de la eritropoyesis y determinar el origen central o
periférico de una anemia, así como para enjuiciar su carácter regenerativo o
arregenerativo.
o Si es patológica
especificar en qué enfermedad (es) se encuentran: Los
valores normales de reticulocitos para el adulto en sangre periférica oscilan
entre 35 y 75 x 10 9 /L DE 0.5% a 2.0%, valores inferiores indican una
eritropoyesis ineficiente.

Eritrocito:
o Tamaño: 2 µ de espesor, con un diámetro de aproximadamente
7 µ.
o Forma: Tiene
forma oval o redondeada, con una depresión o zona más clara en el centro,
cuando se hace un corte transversal tiene una forma de disco bicóncavo,
o Descripción del
núcleo y citoplasma: célula anucleada
cuyo citoplasma está constituido por la hemoblobina que le confiere el carácter
acidófilo.
o Relación
núcleo/citoplasma: No existe núcleo.
o Organelas y/o
inclusiones evidentes en la misma: En condiciones normales es un citoplasma
desprovisto de gránulos o de otro tipo de inclusiones, su tarea principal es
transporte de O2 y CO2 lo que depende de la calidad de la hemoglobina
o Sitio del cuerpo
donde se pueden encontrar normalmente: Sangre
periférica.
o Si es patológica
especificar en qué enfermedad (es) se encuentran: sangre. (Manascero 2,008)
 |
| Recuperado de: http://griho2.udl.es/carles/medicina/sp/eritro.html |
La leucopoyesis, o
desarrollo de leucocitos, con excepción de los linfocitos se produce en el
mismo lugar de la eritropoyesis. Estos sitios cambian a lo largo de la vida,
comenzando con el saco vitelino del mesénquima, seguido por el bazo, y el
hígado, y por último la médula ósea. Este cambio debería considerarse
unidireccional, dado que, con excepción de la metaplasia mieloide, el bazo, y
el hígado no participan en la formación de células sanguíneas después del
nacimiento. Tiene como célula de origen la Célula
madre hematopoyética, y a partir de ella pueden derivarse hacia:
·
La Célula madre mieloide, que al madurar y pasar por diversos estadíos celulares puede dar
origen a dos líneas celulares, la Serie Granulopoyética, que da origen a los
Granulocitos, neutrófilos, eosinófilos y basófilos; o bien madura hacia la
Serie Monocítica, que da origen a los monocitos.
·
La Célula madre linfoide, que da origen a los
Linfocitos B o a los Linfocitos T. (Miale, 2002)
Granulopoyesis Los granulocitos
contienen gránulos visibles y se desarrollan en la médula ósea, en las zonas
más próximas de las trabéculas óseas. Se subdividen de acuerdo con su
morfología y pueden clasificarse en los que contiene gránulos grandes y se
visualizan con facilidad, estos a su vez se dividen de acuerdo con el tipo de
reacción neutrófila, eosinófila y basófila. Los granulocitos pueden encontrarse
en cuatro lugares: en médula ósea, circulando libremente en sangre periférica,
y en los tejidos, estos sitios se denominan compartimientos granulocitos, el
comportamiento de la médula ósea es bastante extenso y posee tres funciones:
proliferación, maduración y almacenamiento. La formación de granulocitos o se
inicia cuando la IL-3 estimula una célula pluripotencial hacia una unidad
formadora de colonias granulomonociitca (UFC-GM) y a partir de ésta se realiza
la diferenciación a mieloblasto, desde mieloblasto a segmentado, demora
aproximadamente entre siete y once días. Las primeras interleucinas descritas
que intervienen en este proceso so la IL-3 en la diferenciación hacia
neutrófilos y la IL-5 en la formación de eosinófilos. Los inhibidores del
crecimiento son, por ejemplo, las prostanglandinas.
Mieloblasto:
o
Tamaño: 15 y 20 µ.
o
Forma: Oval o
redondeada y contorno liso.
o
Descripción
del núcleo y citoplasma: El núcleo es redondo y está provisto de una
cromatina finamente reticulada, con presencia de dos a cuatro nucléolos bien
visibles. El citoplasma es de color basófilo, o hiperbasófilo con
hiperbasofilia, aunque menos intensa que el del pronormoblasto, por lo general
la hiperbasofilia es periférica o moteada. Expresan marcadores específicos de
linaje como MPO y el CD33.
o
Relación
núcleo/citoplasma: Alta, 5:1 a 7:1
o
Organelas
y/o inclusiones evidentes en la misma: La mieloperoxidasa
puede encontrarse en toda la célula, y junto con las enzimas necesarias para el
estadillo de peroxidasa y superoxido. Posee de 2 a 5 nucleolos, bastante
delimitados, redondos u ovales, de color azul pálido, y la cromatina tiende a
ser más clara en las proximidades del nucleolo. Carece de gránulos, pero las
células fagocíticas contiene detritus, hierro, y a veces parasitios.
o
Sitio del
cuerpo donde se pueden encontrar normalmente: Médula ósea, ˃ 5 %,
ausente en condiciones normales
o
Si es
patológica especificar en qué enfermedad (es) se encuentran: Leucemia
mieloide ph/ BCR, leucemia mielomonocítica crónica tipo mieloproliferativa
(LMMC, MP), mielofibrosis idiopática crónica con metaplasia mieloide.
 |
| Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-blanca/ontogenia/promielocito/ |
Promielocito:
o
Tamaño: 6 µ a 25 µ.
o
Forma: Es la
célula de mayor tamaño de la Granulopoyesis normal y su forma es oval o
redondeada.
o
Descripción
del núcleo y citoplasma: El núcleo posee cromatina inmadura, algo más
densa que el mieloblasto, tienen de cero a dos nucléolos y se sitúa en posición
excéntrica. El citoplasma es amplio y basófilo, característicamente contiene un
número variable de gránulos primarios o azurófilos, grandes, gruesos que se
disponen alrededor del núcleo a fin de dejar una zona más clara, agranular, que
corresponde a la zona centrosómica.
o
Relación
núcleo/citoplasma: Menor que el mieloblasto, Moderada, 5:1.
o
Organelas
y/o inclusiones evidentes en la misma: Los gránulos
azurófilos tienen afinidad por los colorantes ácidos, gránulos con tinción
basófila conteniendo estereasa, lisozima, serprocidinas, defensinas, proteínas
aumentadoras de la permeabilidad.
o
Sitio del
cuerpo donde se pueden encontrar normalmente: Médula ósea. ˂ 5%, ausente en sangre en
condiciones normales.
o
Si es
patológica especificar en qué enfermedad (es) se encuentran: Leucemia
promielocitica, enfermedad de Hurler y Scheie.
 |
| Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-blanca/ontogenia/promielocito/ |
Mielocito:
o
Tamaño: 12 a 18 µ.
o
Forma: Redondo u
ovalado.
o Descripción del núcleo y citoplasma: última célula del compartimiento de la médula ósea que puede sufrir mitosis, exhibe gran variabilidad morfológica; el núcleo puede ser redondeado u ovalado con un lado aplanado cerca del Aparato de Golgi, bien desarrollado, el núcleo presenta una escotadura más evidente; la cromatina nuclear se condensa y los nucléolos por lo general no son visibles. El citoplasma se observa claro o con halo rosado pálido.
o Relación núcleo/citoplasma: Baja o muy baja, 2:1.
o Organelas y/o inclusiones evidentes en la misma: Los gránulos primarios disminuyen y aumentan los gránulos secundarios: lactoferrina, histaminidasa, B-microglobulina, apolactoferrina, y activadores del plasminógeno, catecidinas, lisozimas, colagenasa, vesículas secretoras que contienen fosfatasa alcalinas receptores 1 del complemento citocromo b.
o Descripción del núcleo y citoplasma: última célula del compartimiento de la médula ósea que puede sufrir mitosis, exhibe gran variabilidad morfológica; el núcleo puede ser redondeado u ovalado con un lado aplanado cerca del Aparato de Golgi, bien desarrollado, el núcleo presenta una escotadura más evidente; la cromatina nuclear se condensa y los nucléolos por lo general no son visibles. El citoplasma se observa claro o con halo rosado pálido.
o Relación núcleo/citoplasma: Baja o muy baja, 2:1.
o Organelas y/o inclusiones evidentes en la misma: Los gránulos primarios disminuyen y aumentan los gránulos secundarios: lactoferrina, histaminidasa, B-microglobulina, apolactoferrina, y activadores del plasminógeno, catecidinas, lisozimas, colagenasa, vesículas secretoras que contienen fosfatasa alcalinas receptores 1 del complemento citocromo b.
o
Sitio del
cuerpo donde se pueden encontrar normalmente: Constituye menos del
10% de la médula ósea, ausente en sangre en condiciones normales.
o
Si es
patológica especificar en qué enfermedad (es) se encuentran: leucemias.,
agudas.
Mielocito Neutrofilo: Nucleo redondeado, abundante citoplasma
gris-marrón con gránulos.
 |
| Recuperado de http://amunevar.wix.com/hematologia#!galeria-granulocitos: |
 |
| Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-blanca/ontogenia/mielocito-eosinofilo/ |
Mielocito Basófilo: Nucleo redondeado, abundante citoplasma
cubierto con granulaciones azules.
 |
Recuperado de: http://www.studydroid.com/index.php?page=viewPack&packId=107288
|
Metamielocito:
o
Tamaño: 10 y 18 µ.
o
Forma: Oval o redondeada,
o Descripción
del núcleo y citoplasma: Núcleo arriñonado, con una escorbatura
evidente violeta y algo excéntrico, la parte convexa situada en la periferia
celular y la cóncava dirigida hacia el centrosoma. citoplasma de color rosado,
la cromatina se observa condensada y densa, la paracromatina es escasa pero
diferenciable.
o
Relación
núcleo/citoplasma: Discretamente abundante casi 1.5:1
o Organelas
y/o inclusiones evidentes en la misma: El citoplasma posee
una coloración completa de gránulos primarios, y secundarios por lo que
destruye y degrada tóxicos infecciosos o extraños, sin embargo la célula
todavía es incapaz de responder a los factores quimio tácticos e iniciar la
fagocitosis; no se observar nucleolos
o
Sitio del
cuerpo donde se pueden encontrar normalmente: 2 a 5 % en la sangre periférica, aunque esta
proporción puede aumentar, como esta célula todavía no es funcional por
completo, se considera parte del componente de maduración de la médula ósea.
o
Si es
patológica especificar en qué enfermedad (es) se encuentran: Infecciones
o leucemias.
Metamielocito Neutrófilo: Núcleo arriñonado, excentrico, abundante
citoplasma con gránulos rosados y azulados.
 |
Recuperado
de: http://www.telmeds.org/wp-content/uploads/2009/05/metamielocito-neutrofilo.jpg
|
Metamielocito Eosinófilo: Núcleo arriñonado, excentrico, abundante
citoplasma con granulaciones anaranjadas.
 |
Recuperado de: http://www.telmeds.org/wp-content/uploads/2009/05/metamielocito-eosinofilo1.jpg
Metamielocito Basófilo: Nucleo arriñonado, excentrico, abundante
citoplasma con granulaciones azules.
 |
Recuperado de: http://www.studydroid.com/index.php?page=viewPack&packId=107288
Banda o cayado:
o
Tamaño: 10 a 15 µ.
o
Forma: Ovalada.
o
Descripción
del núcleo y citoplasma: El núcleo experimenta ulterior condensación,
que origian una banda en forma de salchicha o cayado, puede estrecharse en uno
o más puntos, pero se observa considerablemente cantidad de cromatina es burda
y de color púrpura azul oscuro. El citoplasma es abundante, azul pálido o rosa,
o
Relación
núcleo/citoplasma: 1: 2
o Organelas
y/o inclusiones evidentes en la misma: Produce gránulos
terciarios conteniendo lisozima y gelatinasa, paracromatina es escasa y el
nucléolo es invisible.
o Sitio del
cuerpo donde se pueden encontrar normalmente: La sangre periférica
puede contener de 1 a 10 % de bandas.
o
Si es
patológica especificar en qué enfermedad (es) se encuentran: Leucemias.
Neutrófilo cayado: Núcleo alargado y
curvado en forma de C o S violeta, y más central, citoplasma más abundante con
gránulos pardos
 |
Recuperado de:
http://bh-lab.blogspot.com/2013_02_01_archive.html
Eosinófilo cayado: Núcleo alargado en
forma de C de color violeta central, abundante citoplasma repleto de gránulos pardo-anaranjados.
 |
Recuperado de : http://www.telmeds.org/atlas/hematologia/serie-blanca/ontogenia/metamielocito-eosinofilo/
Basófilo cayado: Núcleo alargado y
curvado forma de C violeta y central, citoplasma con gruesos gránulos azul
intenso.
 |
| Recuperado de http://hematologiamcs.blogspot.com/ |
Granulocitos
Segmentados:
Neutrofilo Segmentado:
o
Tamaño: 10 a 12 µ.
o
Forma: Redondeada.
o
Descripción
del núcleo y citoplasma: Reconocido por la segmentación del núcleo,
con dos o cuatro lóbulos unidos por un filamento de cromatina delgado, su
cromatina condensada y se tiñe de color morado oscuro, citoplasma rosado con
granulaciones
o
Relación
núcleo/citoplasma: Abundante relación núcleo citoplasma
aproximadamente 1:3 de color rosa o lila
o
Organelas
y/o inclusiones evidentes en la misma: Cromatina burda y
densa que se tiñe de color púrpura azul oscuro, con escasa paracromatina,
o
Sitio del
cuerpo donde se pueden encontrar normalmente: En sangre periférica
de un 40 a 75%
o
Si es
patológica especificar en qué enfermedad (es) se encuentran: Infecciones bacterianas.
 |
Recuperado de: http://ameribalabclinico.blogspot.com/2010/11/neutrofilo-segmentado.html
|
Eosinofilo Segmentado:
o
Tamaño: 10 a 12 µ
o
Forma : Redondeada.
o
Descripción
del núcleo y citoplasma: Núcleo violeta y bilobulado, citoplasma
repleto de gránulos naranja.
o
Relación
núcleo/citoplasma: Abundante relación núcleo citoplasma
aproximadamente 1:3 de color rosa o lila
o
Organelas
y/o inclusiones evidentes en la misma: Fosfatasa ácida,
arisulfatasa, granulos secundarios: proteína básica mayor (MBP), proteína
catiónica eosinófila (ECP), neurotroxina, derivada del eosinófilo (EDN) y la peroxidasa eosinofílica (POE).
o
Sitio del
cuerpo donde se pueden encontrar normalmente: En sangre periférica
de un 0-7%. Se encuentran distribuidos en varios órganos pero prefieren
aquellos que interactúan con el ambiente externo, como los tractos respiratorio
bajo, gastrointestinal y genitourinario, una vez secuestrados pueden sobrevivir
por 6 a 8 días, pudiendo extenderse por semanas bajo efecto de citoquinas,
o
Si es
patológica especificar en qué enfermedad (es) se encuentran: Enfermedades
parasitarias, fenómenos alérgicos. (Miale, 2002)
 |
| Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-blanca/granulocitos-segmentados-maduros/eosinofilo-maduro-bilobulado/ |
Basófilo segmentado:
o
Tamaño 10 a 12 µ
o
Forma: Redonda.
o Descripción
del núcleo y citoplasma: Núcleo con lobulaciones citoplasma con
abundantes gránulos azules que cubren el núcleo, la cromatina densa, posee
generalmente dos o tres lóbulos unidos por puentes cromatínicos, en ocasiones
difíciles de visualizar dad la presencia de las numerosas granulaciones
basófilas propias de la célula.
o
Relación núcleo/citoplasma:
Abundante
relación núcleo citoplasma aproximadamente 1:3 de color rosa o lila
o Organelas
y/o inclusiones evidentes en la misma: Histamina, heparina,
glucógeno y determinados enzimas (peroxidassa), otra enzima es la omega
exonucleasa, localizada en la zona intercelular del basófilo, no tienen
cloroacetoestereasa, ni fosfatasa alcalina,
o Sitio del cuerpo donde se
pueden encontrar normalmente: En sangre periférica
de un 0-2%.
o Si es patológica especificar
en qué enfermedad (es) se encuentran: Reacciones
de hipersensibilidad e inmunidad antiparasitaria. (Miale, 2002)
 |
Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-blanca/granulocitos-segmentados-maduros/
|
MONOPOYESIS: Es
el proceso de formación de los monocitos, este proceso tiene lugar en la médula
ósea hematopoyética en condiciones normales, en condiciones patológicas pueden
formarse en el bazo, al igual que la línea Eritroide, la megacariocitica y la
granulocítica, provienen de una célula madre multipotencial mieloide, que da
lugar a la célula madre unipotencial para los monocitos la cual se divide y da
lugar al monoblasto. Los factores de regulación de la monopoyesis son el factor
estimulador de colonias granulo-monociticas, (CSF-GM) y las necesidades del
orgnaismo.
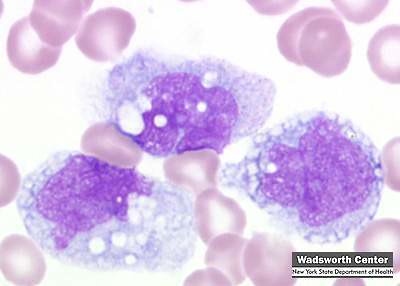
Monoblasto:
o Tamaño: 14 a 18 µ
o Forma : Redonda
o Descripción del
núcleo y citoplasma: Núcleo redondo u
oval membrana nuclear fina y cromatina fina y delicada, paracromatina abundante
netamente delimitada y de color rosa o azul pálidos; citoplasma basófilo
homogéneo, no granulado.
o Relación
núcleo/citoplasma: 6:1
o Organelas y/o
inclusiones evidentes en la misma: De
uno a dos nucléolos, no existe gránulos. (Miale, 2002)
 |
| Recuperado de. https://www.flickr.com/photos/hematologia/3668525599/?rb=1 |
Promonocito:
o Tamaño: 14
a 18 µ
o Forma: Redonda.
o Descripción del
núcleo y citoplasma: Moderadamente
invaginado, membrana fina y delgada, cromatina es fina, lo cual le confiere al núcleo
un aspecto pálido, en comparación con otras células, en acúmulos tosocos,
paracromatina dispersa, mal desaminada, citoplasma moderadamente abundante gris
azul y opaco escasos gránulos de color rosa extraordinariamente finos.
o Relación
núcleo/citoplasma: 5:1
o Organelas y/o
inclusiones evidentes en la misma: De
0 a 1 nucléolo, granulación rosa muy fina, denominada “polvo azurófilo”, sin embargo la demostración
de estos gránulos requiere de una buena técnica de tinción.
 |
| Añadir leyenda: http://zl.elsevier.es/es/revista/revista-laboratorio-clinico-282/clasificacion-las-leucemias-agudas-mieloides-13154845-revision-2010 |
Monocito:
o Tamaño: 12
a 18 µ
o Forma : Redondo.
o Descripción del
núcleo y citoplasma: Núcleo inviginado o
plegado delicado, débilmente teñido, cromatina fina con mucha paracromatina,
citoplasma gris o gris azulado, gránulos muy teñidos muy finos color rosa.
o Relación
núcleo/citoplasma: 4:1
o Organelas y/o
inclusiones evidentes en la misma: por
lo general ninguno, gránulos finos y numerosos como polvo de color lila, tienen
vacuolas de fagocitosis y cuerpos residuales, y algunos sideromas (acúmulos de
ferritina/hierro).
o Sitio del cuerpo
donde se pueden encontrar normalmente: Los
monocitos circulan de dos a tres días en la sangre periférica, pasan por los
tejidos y se convierten en macrófagos tisualres y los histiocitos. (Rodak, 2002)
 |
| Añadir leyenda: Recuperado de: https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEjxSPdwSP4G5dd_4JkTsOUtF42VIg4tBJSTUJlDcQZen9ayLsjt1DrMVBAxvsCGeiQobmYHz42q8sQ0pN1y3tM7zvzKxsKROdK4Ih03qcRPNvg5L-dra2fHltt3uQ2vXhmsoQoAk5srbCE/s1600/monocito.jpg |
LINFOPOYESIS: Es el proceso de formación de los linfocitos
(linfocitos B y T), este tiene lugar no solo en la médula ósea hematopoyética sino
también en tejidos denominados órganos linfáticos primarios (timo y médula
ósea) y en secundarios, (bazo, placas de Peyer, en el tracto gastrointestinal,
anillo de Wadermyer en las amígdalas y adenoides y ganglios linfáticos). La
célula madre hematopoyética pluripotencial (STEM CELL), se divide en la célula
madre hematopoyética de la serie linfoide que da lugar a:
Célula madre unipotenical para linfocitos B (CFU-LB)
Célula madre unipotencial para linfocitos T (CFU-LT)
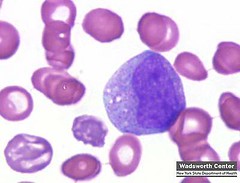
Linfoblasto:
Linfoblasto:
o Tamaño: 10
18 µ
o Forma : Redondo
o oval
o Descripción del
núcleo y citoplasma: Núcleo redondo u
oval, membrana nuclear, clara cromatina bandas finas de color rojo o morado,
citoplasma homogéneo y moderadamente basófilo, a menudo presenta una zona
perinuclear clara.
o Relación
núcleo/citoplasma: 5:1 a 7:1
o Organelas y/o
inclusiones evidentes en la misma: A
menudo, pero no siempre presenta una zona perinuclear, no existen gránulos, y
presenta de 1 a 2 nucléolos, fosfatasa ácida, algunas veces presenta depósitos
de glucógeno .
o Sitio del cuerpo
donde se pueden encontrar normalmente: Médula
ósea.
 |
Recuperado de: http://flickrhivemind.net/User/dione1986/Timeline
|
Prolinfocito:
o Tamaño: 10
a 18 µ
o Forma : Redondo
u oval
o Descripción del
núcleo y citoplasma: Núcleo redondo,
ligeramente invaginado, la cromatina puede ser fina o ligeramente burda de
color púrpura, para cromatina poco clara no acostmbra estar bien delimitada
como el linfoblasto; citoplasma extendido moderadamente basófilo, homogéneo.
o Relación
núcleo/citoplasma: 5:1
o Organelas y/o
inclusiones evidentes en la misma: Generalmente
presenta nucléolos uno, redondo, azul y netamente delimitado, a veces posee
gránulos azurófilos.
o Sitio del cuerpo
donde se pueden encontrar normalmente: Médula
osea.
 |
Recuperado http://amunevar.wix.com/hematologia#!galeria
|
Linfocito:
o Tamaño: Pequeño
de 6 a 18 µ; mediano, de 8 a 14 µ ;
grande de 8 a 18 µ . Tamaño variable principalmente en función de la cantidad
de citoplasma,
o Forma :
o Descripción del
núcleo y citoplasma: Núcleo redondeado u
oval, ligera o profundamente hendido, la membrana nuclear es densa, la
cromatina en acúmulos, tosocos, paracromatina dispersa mal delimitada de color
entre azul pálido y rosa, suele describirse cómo “borrosa”; citoplasma de color
azul celeste o azul claro.
o Relación núcleo/citoplasma:
5:1, 2:1
o Organelas y/o
inclusiones evidentes en la misma: No
contiene nucléolos, citoplasma con gránulos azurófilos que se distinguen con
mayor claridad, tanto el linfocito T como B presenta un apolimerasa de DNA,
desoxinucleotil transferasa terminal.
o Sitio del cuerpo
donde se pueden encontrar normalmente: El
linfocito grande es el más raro en sangre periférica, si maduran en el timo se
consideran linfocitos T, y si maduran en la médula ósea son considerados
linfocitos B,
 |
| Recuperado de: http://www.4shared.com/all-images/_E1lG95w/_Atlas_.html |
TROMBOPOYESIS:
De
igual forma que los eritrocitos y los leucocitos, los megacariocitos se
desarrollan a partir de una célula de la línea mieloide. La trombopoyetina se
genera sobre todo en el riñón, y en menor medida en el hígado y en el bazo y en
la respuesta a la demanda de plaquetas, esta se une de manera específica al
receptor de trombopoyetina C-mply estimula el crecimiento de megacariocitos y
la producción de plaquetas.
Mecarioblasto:
o Tamaño: 25
a 35 µ
o Forma : Redondeada.
o Descripción del
núcleo y citoplasma: Núcleo redondo u
oval; grande; con cromatina delicada que se tiñe de color púrpura y escasa
paracromatina, citoplasma escaso, irregularmente basófilo aveces con extrusiones citoplasmáticas.
o Relación
núcleo/citoplasma: 10:1
o Organelas y/o
inclusiones evidentes en la misma: De
2 a 6 nucleolos pequeños y mal delimitados
o Sitio del cuerpo
donde se pueden encontrar normalmente: Médula
ósea.
 |
| Recuperado de http://plaquetas.wordpress.com/atlas/ |
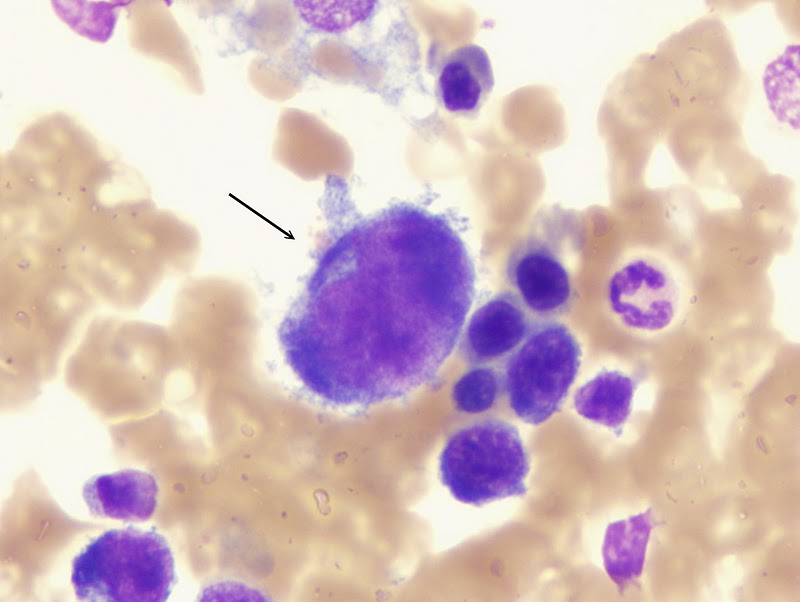
Promegacarioblasto:
o Tamaño: 25
a 50 µ
o Forma: Redondeada.
o Descripción del
núcleo y citoplasma: Núcleo grande e
irregular, más tosco que el del megacarioblasto, puede parecer lobulado, aveces
existen dos o más núcleos diferenciados, citoplasma moderadamente basófilo con
cierta policromasia.
o Relación
núcleo/citoplasma: 10:1
o Organelas y/o
inclusiones evidentes en la misma: Nucléolos
son difíciles de ver y hay de ninguno a dos. Contiene unos cuantos gránulos
azurófilos finos en la zona perinuclear. En algunas circunstnacias se produce
una formación precoz de plaquetas en la periferia.
o Sitio del cuerpo
donde se pueden encontrar normalmente: Médula
ósea.
 |
| Recuperado de: http://plaquetas.wordpress.com/atlas/ |
Megacariocito:
o Tamaño: La
mayor célula sanguínea, de 40 a 100 µ
o Forma : Varía
en su aspecto así que no es posible definir un asola forma típica.
o Descripción del
núcleo y citoplasma: Núcleo multiforme,
generalmente parece un cálculo coraliforme; la cromatina es tosca e
irregularmente acumulada. Abundante citplasma, pálido
o Relación
núcleo/citoplasma: Normalmente nula.
o Organelas y/o
inclusiones evidentes en la misma: Nucléolos,
ninguno o ambos casis siempre invisibles, sin embargo en células asiladas cabe
observar muchos nucléolos endomitóticas poliploides del núcleo, gránulos
azurófilos finos que se distribuyen de forma regular y dispersa o en acúmulos.
Puede presentar proyecciones citoplasmáticas parecidas a seudópodos, que
originan plaquetas.
 |
| Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-trombocitica/plaquetas/ |
Plaquetas:
o Tamaño: 2.5
µ
o Forma : Discos
leniformes con márgenes lisos.
o Descripción del
núcleo y citoplasma: Tiene un área
periférica formada por la membrana plaquetaria, que contiene glucoproteínas,
unidas a la membrana una cubierta externa denominada glucocálix y un
citoesqueleto rico en filamentos de actina y miosina y microtúbulos. Cromómero:
con gránulos alfa, y proteínas de adhesión.
o Relación
núcleo/citoplasma: Nula.
o Organelas y/o
inclusiones evidentes en la misma: Fosfatasa
ácida, fibirnóngeno, factores de crecimiento plaquetario, factor neutralizante
de heparina, proteínas de adhesión glucoronidasa, trombospondina.

o Sitio del cuerpo
donde se pueden encontrar normalmente: Sangre
periférica.
Microesferocitos: Constituyen en realidad, formas preliticas que se originan por recorte simétrico o parcial de la membrana de hematíe. Son células con una disminución significativa de la relación superficie / volumen y un aumento de la concentración media de la hemoglobina corpuscular (PCHC), factores que como se ha dicho determinan la deformabilidad del hematíe. Los microesferocitos, se observan en anemias hemolíticas heredadas como en la SH. Se asocia también, con desordenes que cursan con formación de Cuerpos de Heinz. El mecanismo esferogénico, en este caso, al igual que en la hemólisis inmune es la fagocitosis parcial de porciones de la célula que contiene agregados de hemoglobina desnaturalizada y porciones de membrana sensibilizada respectivamente. La injuria térmica (quemaduras mayores) y la injuria mecánica induce hemólisis intravascular, fragmentación celular y formación de microesferocitos. La hipofosfatemia severa cursa como una anemia hemolítica caracterizada por una marcada microesferocitosis
LEUCEMIAS
Etiopatogenia
• La PV es un SMPc de etiología desconocida que se origina de un progenitor hematopoyético pluripotencial. Está caracterizado por una producción anormal de eritrocitos, leucocitos y plaquetas en ausencia de un estímulo fisiológico reconocible.
Cuadro clínicoAunque no es una enfermedad eminentemente mortal, sí afecta la calidad de vida de los pacientes que la padecen. Las alteraciones clínicas y de laboratorio que pueden verse son:
• Leucocitosis persistente.
• Trombocitosis persistente.
• Microcitosis por déficit de Fe.
• Esplenomegalia.
• Prurito generalizado (posterior al baño).Manual de Prácticas Médicas - Hospital Hermanos Ameijeiras 20
• Trombosis inusuales.
• Eritromelalgia.
Las complicaciones más graves en la PV incluyen la trombosis y las hemorragias, principales causas de morbimortalidad. Las trombosis se producen tanto en la microcirculación, como en los grandes vasos. Hasta 20 % de los pacientes puede tener algún episodio trombótico:
• AVE
• ICT
• Oclusión de la vena o arteria de la retina,
• Isquemia coronaria,
• TEP,
• Trombosis de la vena portal o hepática,
• Trombosis venosas profundas
• Isquemias digitales
Su incidencia depende de la edad del paciente, la historia anterior de trombosis y la presencia de factores de riesgo aterosclerótico. Las complicaciones hemorrágicas son menos frecuentes y su aparición está casi siempre relacionada con el uso de AINES y el ASA.
La transformación de la enfermedad en una LMA es una complicación posible que puede presentarse, llevando al paciente a una evolución fatal.
Diagnóstico: Debe distinguirse entre una eritrocitosis secundaria, dependiente de la eritropoyetina (Epo) y la que no depende de esta y que provoca un aumento del volumen globular eritroide absoluto.
•No existen marcadores histológicos ni clonales disponibles que identifiquen inequívocamente a la PV.
• Las anormalidades citogenéticas están presentes en menos de 30 % de los pacientes al diagnóstico y tampoco son específicos.
• Además la BMO y su histología pueden ser normal o indistinguibles de la MPy la TE.
Características clínicas
• En ocasiones el paciente acude al médico por notarse el mismo una masa en el abdomen o por síntomas atribuibles a la esplenomegalia como molestias o dolor. Pueden ocurrir infartos esplénicos, periesplenitis o hematomas subcapsulares que provocan dolor intenso en el cuadrante superior izquierdo del abdomen y/o en el hombro de ese lado. Otros síntomas relacionados con la esplenomegalia son las diarreas por compresión del colon, la sensación precoz de llenura con los alimentos por compresión gástrica y los edemas en miembros inferiores.• El paciente puede manifestar síntomas de anemia y síntomas generales como astenia, perdida de peso, sudores nocturnos, febrícula, que pueden ser consecuencia de un estado de hipercatabolia. Pueden observarse litiasis renal por uratos o un cuadro de gota.
• El paciente puede manifestar síntomas relacionados con la localización de la hematopoyesis extramedular: hemorragias digestivas, compresión de médula espinal, convulsiones focales, síntomas de tumor cerebral, ascitis, derrame pericárdico, derrame pleural, hemoptisis y fallo respiratorio.
• Puede presentarse un cuadro completo de hipertensión portal por un flujo esplenoportal marcadamente incrementado. Puede ocurrir una trombosis portal o hepática como complicación.
• La leucopenia se observa en menos de 25 % de los casos, mientras que la leucocitosis es frecuente, aunque es raro observar cifras mayores de 50 x109/L. Predominan los granulocitos maduros, pero se observan también formas jóvenes (mielocitos, juveniles, etc.), y un número pequeño de blastos y células de Pelger Huet.
• La trombocitosis es mas frecuente que la trombocitopenia y puede detectarse en el 35-50 % de los casos. La trombocitopenia se incrementa a medida que avanza la enfermedad. En la extensión pueden observarse megacariocitos, fragmentos de megacariocitos y plaquetas gigantes.
• En los estudios de la hemostasis es frecuente la disfunción plaquetaria y se puede detectar una CID, clínicamente silente, en 15 % de los casos
·
Linfoma
de Hodgkin con esclerosis: nodular: el
linfoma de Hodgkin con esclerosis nodular es la forma más frecuente de LHc;
hasta el 80% de las personas con LHc tienen este tipo. Aparece más
frecuentemente en adultos jóvenes, en especial, mujeres. Además de las células
de Reed-Sternberg, hay franjas de tejido conectivo en el ganglio linfático. A
menudo este tipo de linfoma compromete los ganglios linfáticos en el mediastino
(tórax).
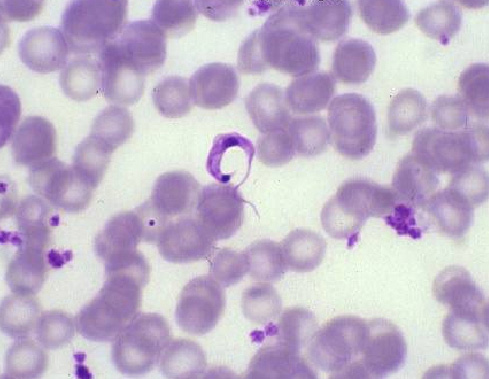
MALARIA:
El paludismo es causado por el protozoo del género Plasmodium. Se han identificado alrededor de 150 especies pero son de relevancia las especies que se han adaptado al hospedero humano y son transmitidas principalmente por un vector anofelino:Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae y Plasmodium ovale. Plasmodium knowlesi, parásito de ciertos macacos en el sudeste de Asia, también se ha identificado como causa de enfermedad. (Cox-Singh J. 2012; Sermwittayawong et al., 2012).

Microgametocito:
La enfermedad de Chagas se origina a partir de la infección con el parásito protozoario Trypanosoma cruzi, miembro de la familia tripanosomatidae . La mayoría de las cepas de este parásito se pueden clasificar en 2 grupos principales, T. cruzi I y T. cruzi II, que incluso se pueden dividir en diversos linajes (p. ej., T. cruzi IIa). Los linajes tienden a asociarse a determinadas especies de huéspedes, aunque esta relación no es absoluta.
Transmisión y ciclo de vida
La fase aguda se define como el período durante el cual los parásitos se pueden encontrar fácilmente en la sangre. Muchas personas, especialmente los adultos, son asintomáticas durante esta etapa. Los síntomas de la fase aguda son sumamente variables y pueden incluir fiebre, dolor de cabeza, anorexia, malestar, mialgia, dolor en las articulaciones, debilidad, náuseas, vómitos, diarrea, hepatomegalia, esplenomegalia y linfadenopatía generalizada o localizada. En algunos casos puede ocurrir edema, ya sea generalizado o localizado, en el rostro o en las extremidades inferiores. En ocasiones, se observa un chagoma (un endurecimiento localizado indoloro) en el lugar de la piel por donde el parásito ha ingresado. Si el ingreso se produce a través de las membranas mucosas oculares, se puede producir un edema indoloro de uno u ocasionalmente ambos ojos, con frecuencia acompañado de conjuntivitis y aumento de tamaño de los ganglios linfáticos locales. Este síndrome, que también se denomina signo de Romaña, generalmente persiste durante 1 ó 2 meses. Los pacientes ocasionalmente desarrollan un sarpullido, pero este generalmente desaparece luego de varios días. En la mayoría de los casos, los signos clínicos se mejoran en semanas o meses sin tratamiento; sin embargo, algunos casos agudos pueden ser mortales.
TOXOPLASMOSIS:
El cinetoplasto es una subestructura de la gran mitocondria, con DNA único y se encuentra asociado estrechamente al bolsillo flagelar y al cuerpo basal del flagelo. La presencia del cinetoplasto da el nombre al grupo de protozoos incluidos en el orden Kinetoplastida.
Los promastigotes metacíclicos, extracelulares, una vez en la probóscide del
mosquito hembra, también conocido como "mosca de arena", son
introducidos en la piel de un hospedero vertebrado durante la ingesta de
sangre. Los parásitos son fagocitados en piel por macrófagos, células de Langerhans
y activan el complemento. Aunque muchos promastigotes son destruidos por los
polimorfonucleares, algunos se transforman en amastigotes en las células del
sistema fagocítico mononuclear; en los fagolisosomas (vacuola parasitófora),
pierden el flagelo y se transforman en amastigotes, multiplicandose por
división binaria. La replicación ocurre en cantidades que oscilan desde decenas
hasta cientos. Las células infectadas se rompen finalmente y los amastigotes se
diseminan, de acuerdo a factores del parásito y del hospedero, entre otros,
hacia diferentes tejidos. Cuando moscas libres de infección se alimentan de
individuos infectados, ingieren las células con amastigotes que sufren cambios
bioquímicos y morfológicos en el intestino medio del insecto, se multiplican y
finalmente migran a la probóscide como promastigotes metacíclicos, altamente
infectantes y promastigotes.
Referencias Bibliográficas:
 |
| Recuperado de: http://www.telmeds.org/atlas/hematologia/serie-trombocitica/plaquetas/ |
ANORMALIDADES
Anormalidades Eritrocitarias:
ALTERACIONES EN EL COLOR:
Hipocrómicos:
·
Tamaño: mayor de 1/3 del diámetro de la
célula.
·
Forma :
·
Descripción del núcleo y citoplasma: Son eritrocitos muy poco
hemoglobinizados con una exagerada de palidez.
·
Relación núcleo/citoplasma:
·
Organelas y/o inclusiones evidentes en la misma:
·
Sitio del cuerpo donde se pueden encontrar
normalmente: Sangre periférica.
·
Si es patológica especificar en qué enfermedad (es) se
encuentran: Las células
hipercrómicas son el resultado de la disminución o deterioro de la síntesis de
la hemoglobina, se relaciona con anemia por deficiencia de hierro, anemia
sideroblástica, talasemias y anemias de enfermedades crónicas.
 |
| Recyoeradi de; http://bhematica.blogspot.com/2010/04/hipocromia-consiste-en-la-existencia-de_19.htm |
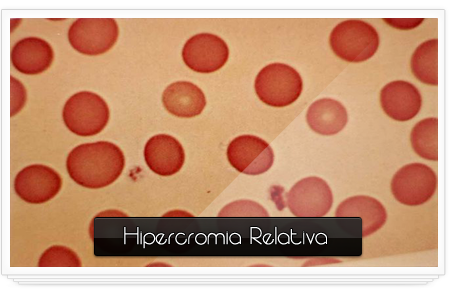
Hipercrómicos:
·
Tamaño: mayor de 1/3 del diámetro de la célula.
·
Forma:
·
Descripción del núcleo y citoplasma: Son eritrocitos
intensamente coloreados.
·
Relación núcleo/citoplasma:
·
Organelas y/o inclusiones evidentes en la misma:
·
Sitio del cuerpo donde se pueden encontrar
normalmente: Sangre periférica.
·
Si es patológica especificar en qué enfermedad (es) se
encuentran: Son el resultado de alteraciones el grosor de la célula y disminución de
la presión central y no la concentración media de la hemoglobina es
más predominante en los megalitos pero sobre todo
en la esferocitosis hereditaria.
 |
| Recuperado de: http://misangre4b.blogspot.com/2013_04_01_archive.html |
Policromatófilos:
·
Tamaño: : Suelen ser más grandes que las
células normales, se manifiesta por el color azul grisáceo con la coloración de
Romanowsky, el tinte azuloso es producido por la presencia De RNA residual en
el citoplasma.
·
Forma:
·
Descripción del núcleo y citoplasma: Coloración grisácea con Romanowsky
·
Relación núcleo/citoplasma:
·
Organelas y/o inclusiones evidentes en la misma: Residuos de RNA residual en el citoplasma.
·
Sitio del cuerpo donde se pueden encontrar
normalmente: Sangre periférica.
·
Si es
patológica especificar en qué enfermedad (es) se encuentran: Se presenta en pacientes cuando hay
disminución en la supervivencia del eritrocito (hemólisis, hemorragia).
 |
| Recuperado de; http://www.talassemias.com.br/talassemiastipos/tal-alfa.htm |
ALTERACIONES EN EL
TAMAÑO:
ANSOCITOSIS:
Cuando patológicamente se aumenta o disminuye
dicho diámetro el resultado será:
·
Macrocitos:
Son eritrocitos con diámetro superior a 8.5 micras. La macrocitosis está
usualmente acompañada de un PVC por encima de 100 fL. Su aparición puede estar
asociada a: Deficiencia de vitamina B12 y/o ácido fólico. Estrés medular cuando
hay eritropoyésis acelerada. Trastornos hepáticos. Es importante anotar que
normalmente los extendidos de sangre periférica de neonatos se caracterizan por
presentar una macrocitosis significativa.
 |
| Recuperado de http://www.telmeds.org/atlas/hematologia/serie-roja/anomalias-de-tamano-anisocitosis/macrocitos/ |
·
Microcitos:
Recibe este nombre el glóbulo rojo que presenta un diámetro inferior a 6.0
micras, la microcitosis está usualmente acompañada de un PVC por debajo de 75
fL. Se observan microcitos en entidades que cursan con alteraciones
cuantitativas de la hemoglobina como son las anemias por deficiencia de hierro
y las talasemias. Generalmente los microcitos se acompañan de bajo contenido de
hemoglobina por lo cual se denominan microcitos hipocrómicos.
 |
| Recuperado de http://www.telmeds.org/atlas/hematologia/serie-roja/anomalias-de-tamano-anisocitosis/microcitos/ |
·
Megalocitos.
Son los “grandes macrocitos ovales”, células donde se combina una alteración
del tamaño y de la forma, pueden llegar hasta tener 12 micras de diámetro.
Característicamente se observan en anemias megaloblásticas
 |
| http:// |
VARIACIÓN EN LA FORMA (POIKILOCITOSIS):
·
Equinocito:
Característicamente aparece como una célula dentada que presenta sobre su
superficie pequeñas progresiones redondeadas a manera de bombas o vesículas de
tamaño uniforme y simétricamente distribuidas que con el MEB, semejan un erizo
de mar.
Los equinocitos
se observan en pacientes con: Uremia, defectos del metabolismo glicolítico
(deficiencia de Piruvato Kinasa) y en algunos pacientes con anemia
microangiopática. Son comunes en neonatos y se observan transitoriamente
después de transfusiones masivas de sangre almacenada (lesión de
almacenamiento) y caracterizan el agotamiento metabólico de la célula roja
senil.
 |
| Recuperado de http://www.iqb.es/hematologia/atlas/equinocitos/equinocito.htm |
·
Acantocito:
Al microscopio electrónico esta célula se observa como una estructura densa
e irregularmente contraída. Bajo el microscopio de luz es un hematíe con
escasas proyecciones espiculadas que no presentan una distribución homogénea y
varían en longitud y número. Los acantocitos se generan cuando las células
rojas normales se exponen a condiciones que modifican el contenido lipídico de
su membrana, alterando la relación colesterol libre / fosfolípidos en ciertas
zonas de membrana. Una vez producida esta forma, es irreversible.
Los acantocitos
también puede observarse en cirrosis alcohólica con anemia hemolítica, en
algunos casos de deficiencias de Piruvato Kinasa, en hepatitis del recién
nacido y después de la esplenectomía.
 |
| Recuperado de http://www.iqb.es/hematologia/atlas/acantocito/acantocito.htm |
·
Estomatocito:
En los individuos normales el 3% o menos de las células rojas en el
extendido de sangre periférica son estomatociticas. El estomatocito se define
como una célula unicóncava que en los extendidos coloreados con Wright presenta
una depresión central elongada con apariencia de boca o estoma (de allí su
nombre) que sustituye el área de palidez central redondeada de los discos
bicóncavos. Es un estado transicional en la transformación
discocito-esferocito.
El estomatocito de
la estomatocitosis hereditaria se genera por una falla en la bomba de Na+ - K+.
Cuando la entrada de Na+ excede la perdida de K+, la célula roja
progresivamente gana cationes, agua y se hincha. De allí el sinónimo de
HIDROCITO, fenómeno opuesto a la formación de crenocitos o xerocitos
acompañados de deshidratación.
 |
| Recuperado de http://www.iqb.es/hematologia/atlas/estomatocito/estomatocito.htm |
·
Degmacitos:
También llamadas por su aspecto células mordidas, son hematíes en los que
la hemoglobina ha sido desnaturalizada y precipitada, con lo que las células
muestran diferentes defectos. Estas células suelen ser eliminadas por el bazo. Se presentan en: - anemia inducida por fármacos- deficiencia en glucosa-6-fosfato
deshidrogenasa – talasemia -
hemoglobinopatías inestables.
 |
| Recuperado de http://www.iqb.es/hematologia/atlas/degmacito/degmacito.htm |
·
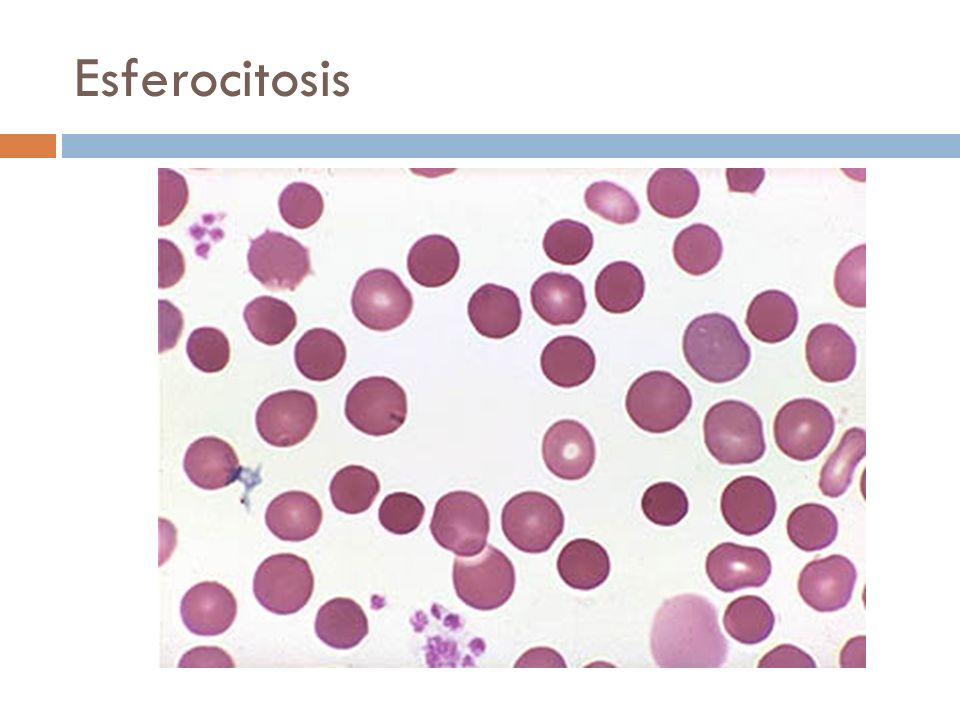
Esferocito:
Hematíes maduros de un diámetro entre 6.1 y 7 mm, esféricos y uniformemente
coloreados. Tienen un volumen algo más pequeño que las células normales y una
concentración mayor de hemoglobina Se
presentan en las siguientes condiciones:
- Esferocitosis hereditaria (Enfermedad de Minkowsky-Chauffard)- Anemias inmunohemolíticas-
Hiperesplenismo - Quemaduras graves- Hipofosfatemia- Septicemia por Clostridium
Welchii
Así dentro
del grupo de los hematíes esféricos se distinguen los siguientes subtipos:
·
Esferocitos de la Esferocitosis Hereditaria (SH):
Su formación se asocia con dos defectos moleculares diferentes: deficiencia
parcial de espectrina (SH [Sp+]) y unión defectuosa de espectrina a la proteína
4.1 (SH [Sp-4.1]). Este desorden genera un esqueleto de membrana inadecuado que
perturba la conservación de la biconcavidad y deformabilidad del glóbulo rojo.
Los esferocitos hereditarios son selectivamente retenidos en la pulpa esplénica
y después de sufrir un número determinado de estos episodios de “manipulación
esplénica” experimentan lisis o fagocitosis en la pulpa roja.
·
Esferocitos con aumento de volumen: Hidrocitos: Se
presentan por una lesión bioquímica de membrana que permite un exceso de
difusión intracelular de sodio y agua. La célula para eliminar el exceso,
estimula el bombeo activo del catión, con un incremento concomitante de la
glicólisis anaerobia y del ATP. El agotamiento energético provoca finalmente la
destrucción celular por el mecanismo conocido como LISIS OSMOTICA COLOIDAL, que
caracteriza la lisis mediada por el complemento y algunas toxinas y venenos.
 |
| Recuperado de http://slideplayer.es/slide/118762/ |
Microesferocitos: Constituyen en realidad, formas preliticas que se originan por recorte simétrico o parcial de la membrana de hematíe. Son células con una disminución significativa de la relación superficie / volumen y un aumento de la concentración media de la hemoglobina corpuscular (PCHC), factores que como se ha dicho determinan la deformabilidad del hematíe. Los microesferocitos, se observan en anemias hemolíticas heredadas como en la SH. Se asocia también, con desordenes que cursan con formación de Cuerpos de Heinz. El mecanismo esferogénico, en este caso, al igual que en la hemólisis inmune es la fagocitosis parcial de porciones de la célula que contiene agregados de hemoglobina desnaturalizada y porciones de membrana sensibilizada respectivamente. La injuria térmica (quemaduras mayores) y la injuria mecánica induce hemólisis intravascular, fragmentación celular y formación de microesferocitos. La hipofosfatemia severa cursa como una anemia hemolítica caracterizada por una marcada microesferocitosis
 |
| Recuperado de http://woomitha12.blogspot.com/2010/09/anormalidades-morfologicas-del.html |
· Dianocito-codocito-“Target
cell”: La verdadera forma circulante es la de campana, en las extensiones
convencionales sufre una redistribución anómala de la hemoglobina asumiendo la
forma de célula blanco en diana (target cell). El codocito es la expresión
morfológica resultante del incremento de la relación superficie / volumen que
bien puede darse por: Expansión de
la superficie, por aumento de los lípidos de membrana, sin cambio en el
volumen, asociado a la enfermedad hepática obstructiva, deficiencia de L.C.A.T.
(Lecitin-Colesterol-Acy 1-Transferasa) y post-esplenectomía. Disminución de
volumen por reducción cuantitativa de la hemoglobina intracelular en entidades
como talasemia y anemia por deficiencia de hierro. Pérdida selectiva de K+ y
agua por defecto de permeabilidad o carencia de ATP (Efecto Gardos). Presencia
de hemoglobinas anormales tales como S, C, D, E. Con excepción de las células rojas deshidratadas, un incremento en
la superficie de membrana, bien sea relativo o absoluto, tiene poco efecto
sobre la deformabilidad o vida media del hematíe y es usualmente inocuo. Mecánicamente se puede inducir la
formación de dianocitos por aplastamiento excesivo; de ahí la necesidad de
observar si la distribución de estas células es uniforme (verdadera) o si por
el contrario se concentra en áreas delgadas del extendido.
 |
| Recuperado de http://biometriahemativa.blogspot.com/2010/04/alteraciones-de-los-eritrocitos.html |
·
Eliptocito
u ovalocito: Es básicamente un disco bicóncavo oval con extremos
redondeados, su forma varía desde una simple distorsión ligeramente oval hasta
casi cilíndrica elongada con polarización de la hemoglobina. En los extendidos
de sangre de individuos normales, las células elípticas u ovales usualmente
constituyen menos del 1% de los eritrocitos. Proporciones un poco más altas se
observan en pacientes con anemias, particularmente megaloblásticas y
microcíticas hipocrómicas. La presencia de un número significativo de células
elípticas (20% - 75%) en sangre periférica se asocia con un desorden clínico y
genéticamente heterogéneo conocido como ELIPTOCITOSIS HEREDITARIA (EH). El
defecto bioquímico de los eliptocitos hereditarios involucra las proteínas de
esqueleto de membrana. Se encuentran
las siguientes condiciones:
- eliptocitosis
hereditaria- anemia ferropénica-anemia mieloptísica- anemia megaloblástica-
talasemia - anemia sideroblástica - anemia congénita diseritropoyética.
 |
| Recuperado de http://galeon.com/hematologiauss/microteca.htm |
·
Célula
falciforme o drepanocito: Es una célula elongada con extremos puntiagudos o
espiculados que semejan una hoz o media luna. La patología básica de la
transformación falciforme está directamente ligada con la concentración de
hemoglobina S (B6 Glu – Val), que tiene la propiedad de polimerizarse en
condiciones de hipoxemia, acidosis, altos niveles de 2-3 DPG y deshidratación
del glóbulo rojo.La polimerización de la desoxi-hemoglobina S, es un proceso
altamente complejo que resulta de la formación de tetrámeros de hemoglobina en
solución. La transformación sol-gel es el hallazgo que permite cambios en la
viscosidad, distorsión en la morfología celular e infartación de órganos que
caracterizan las manifestaciones clínicas de la enfermedad.
 |
| Recuperado de http://bhematica.blogspot.com/2010/04/drepanocitosis-la-anemia-de-celulas.html |
·
Dacriocito
o célula en gotera: Se refiere este término a la célula roja caracterizada
por una elongación única asumiendo una forma periforme o gotera rasgada. Su
génesis permanece oscura. Constituye un hallazgo ocasional en anemias
megaloblásticas, talasemias y mielofibrosis.
También aparecen
estos hematíes en la hematopoyésis extamedular (mielofibrosis, anemia
mieloptísica).
 |
| Recuperado de http://www.fotolog.com/tanianc21/40929791/ |
·
Esquistocito:
Hematíes maduros, de tamaño y formas irregulares o fragmentos celulares,
debidos a roturas de la membrana.Se presentan en:- anemias hemolíticas por
fragmentación mecánica - síndrome hemolítico urémico- púrpura trombótica
trombocitopénica- microangiopatías del embarazo y puerperio.- Coagulación
intravascular diseminada.
 |
| Recuperado de http://www.galenusrevista.com/Purpura-trombotica.html |
·
Queratocito
o célula mordida: Son los hematíes parcialmente fragmentados, producto de
la función “pitting” del bazo, en la que la célula pierde una porción de la
membrana sin alterar el contenido intracelular de la hemoglobina. Al
observarlos coloreados con Wright, en los extendidos de sangre periférica,
presentan proyecciones que semejan cuernos, resultantes de la ruptura de la
vacuola que se forma alrededor del cuerpo de inclusión. Aparecen en entidades
que cursan con la formación de cuerpos de Heinz, particularmente en la deficiencia
de glucosa 6 fosfato deshidrogenasa y talasemias.Se presentan estos hematiés
en: - glomerulonefritis - deficiencia de piruvato kinasa- hemólisis por
fragmentación mecánica intravascular asociada a anormalidades valvulares -
anemia hemolítica microangiopática.
 |
| Recuperado de http://mesa54d.blogspot.com/feeds/posts/default |
·
Punteado
basófilo. Agregados anormales de ribosomas. En síndromes talasémicos,
intoxicación por plomo, deficiencia de hierro, síndromes que se acompañen de
eritropoyésis ineficaz. Coloración Wright.
 |
| Recuperado de http://www.telmeds.org/atlas/hematologia/serie-roja/anomalias-de-contenido/punteado-basofilo/ |
·
Cuerpos
de Howell – Jolly. Remanentes nucleares. En asplenia y estados
hipoesplénicos, anemia perniciosa y anemias severas por déficit de hierro.
Coloración Wright.
 |
| Recuperado de http://www.telmeds.org/atlas/hematologia/serie-roja/anomalias-de-contenido/cuerpos-de-howell-jolly/ |
·
Anillos
de Cabot. Remanentes nucleares en forma de anillos circulares doblados
sobre si mismos o en figura de ocho. En intoxicación por plomo, anemia
perniciosa y anemias hemolíticas. Coloración Wright.
 |
| Recuperado de http://www.basesmedicina.cl/hematologia/15_3_anemia_mega/contenidos.htm |
·
Cuerpos
de Heinz. Hemoglobina agregada o desnaturalizada. En pacientes con
síndromes talasémicos o de hemoglobinas inestables, en el estrés oxidante
cuando hay deficiencias enzimáticas de la vía de pentosas principalmente y
pacientes con asplenia. Coloración azul de cresil brillante.
 |
| Recuperado de http://www.iqb.es/hematologia/atlas/variantes_eritrocitos/cuerpos_heinz.htm |
ALTERACIONES
LEUCOCITARIAS:
·
Basofilia
citoplasmática: se observa como un citoplasma azul difuso. Recordar que
esta característica es normal en las células inmaduras (mieloblasto y
progranulocito), pero se la considera un cambio tóxico cuando lo observamos en
células más maduras. Esta basofilia es debida a la retención de ribosomas y
retículo endoplásmico rugoso y es indicativa de un deterioro en la maduración
celular.
 |
| Recuperado de http://patolvet.wordpress.com/2012/04/10/233/ |
·
Granulaciones
tóxicas: representan las granulaciones primarias observadas en los
promielocitos, las cuales normalmente, al madurar la célula pierden su
capacidad tintorial. Son granulaciones de color rojizo y representan
mucopolisacáridos que son retenidos. Es un
cambio que se observa con mucha frecuencia en caninos.
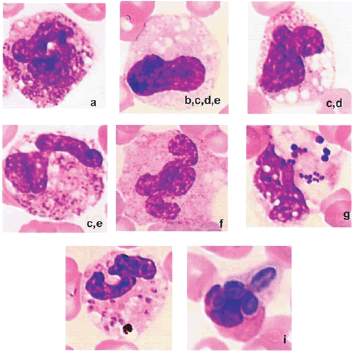 |
| Recuperado de http://www.scielo.org.ar/scielo.php?pid=S0325-29572009000400013&script=sci_arttext |
·
Células
gigantes: son debidas a un asincronismo entre la maduración del núcleo y
el citoplasma. O sea se produce
maduración del citoplasma en el mielocito pero sin división nuclear. En raros casos puede producirse un
doble núcleo. Son células semejantes en apariencia a las células normales pero
con un tamaño aproximadamente el doble de lo normal. Es mucho más frecuente de
observar en felinos.También puede ocurrir que el núcleo comience a madurar con
la formación de una cavidad central: núcleo en anillo.
 |
| Recuperado de http://www.ht.org.ar/histologia/NUEVAS%20UNIDADES/unidades/unidad3/macrof.htm |
·
Cuerpos
de Döhle: son pequeñas áreas de basofilia citoplasmática que resultan de la
agregación aberrante de láminas de retículo endoplásmico rugoso. Son frecuentes
de observar en felinos sin estar asociada a procesos infecciosos o tóxicos, por
ejemplo en una granulopoyesis aumentada.
·
 |
| Recuperar leyenda http://laboratorio-arcano-1.blogspot.com/2012_01_01_archive.html |
ANEMIAS
Anemia ferropriva
La anemia ferropriva y la de las enfermedades
crónicas, o anemia inflamatoria, son las anemias que se observan con mayor
frecuencia en la práctica clínica. La deficiencia de fierro es muy común. En la
actualidad es muy raro observar las características clínicas clásicas de la
deficiencia grave de hierro, como glositis, queilitis, disfagia y coiloniquia;
el reconocimiento clínico de la deficiencia de hierro hoy en día se basa en
determinaciones de laboratorio. Es importante recordar que la deficiencia de
hierro ocurre cuando se produce un desbalance entre las necesidades y el
suministro. Esto puede ocurrir, en primer lugar, debido a demandas no cubiertas
por el hierro absorbido, como ocurre cuando hay aumento de los requerimientos,
dietas demasiado estrictas y en presencia de síndromes de malaabsorción; por
ejemplo, puede haber aumento de requerimientos en niños, mujeres embarazadas,
adolescentes en crecimiento rápido o personas bajo entrenamiento muscular
intenso.
Diagnóstico de anemia ferropriva
El diagnóstico de anemia ferropriva se establece en
presencia de anemia microcítica e hipocroma, es decir, con volumen corpuscular
medio disminuido, habitualmente menor de 80 um3 y hemoglobina corpuscular media
disminuida, que indica menor carga de hemoglobina en los glóbulos rojos. Un
parámetro que se cita con frecuencia, pero se utiliza poco es el ancho de
distribución eritrocitario (ADE), que en la talasemia, la otra gran causa de
anemia microcítica e hipocroma, está disminuido. En la anemia ferropriva el
perfil de hierro se caracteriza por ferremia disminuida, capacidad de fijación
de hierro aumentada y ferritina disminuida. La muestra se debe tomar siempre en
ayunas por la mañana, ya que hay un ritmo circadiano para la ferremia.
Finalmente, lo más importante es el diagnóstico de la causa.
 |
| Recuperado de: http://professoraangela.net/documents/anemia_ferropriva.html |
Anemia
megaloblástica:
En la formación de la síntesis del ADN de las células
precursoras de los hematíes participan tanto la vitamina B12 (cobalmina) como
el ácido fólico. Si alguno de estos componentes falta se produce un descenso en
la velocidad de síntesis del ADN de las células, hecho que conlleva que los
precursores de la serie roja aumenten de tamaño. Asimismo, esta alteración en
la maduración de las células hace que pueda producirse una destrucción de las
mismas, cosa que contribuye a la anemia.Así pues, los hematíes salen en menor
número a la sangre y con un tamaño alterado a causa de esta falta de vitamina
B12 o de ácido fólico, dando lugar a lo que se conoce como anemia
megaloblástica. La falta de ácido fólico es la causa más frecuente de anemia
megaloblástica. El ácido fólico se obtiene de la dieta, a partir de alimentos
como la carne, las legumbres, los frutos secos o las verduras, y se absorbe a
nivel del yeyuno. Se transporta a la médula ósea para intervenir en la síntesis
de ADN de los hematíes y se acumula en depósitos en el hígado. La principal causa de falta de
ácido fólico es su déficit en la dieta, sobre todo por desnutrición y
alcoholismo, aunque también puede ser debido a alteraciones en su absorción y
almacenamiento causadas por diversas enfermedades intestinales y
hepáticas, a situaciones que aumenten su consumo, como el embarazo o ciertos
tumores, o a algunos fármacos como el metrotexate, el trimetoprim o el
triamterene. La vitamina B12 se absorbe a nivel del intestino
delgado tras unirse en el estómago a una proteína que se llama factor
intrínseco. Pasa a la sangre y se transporta unida a otra proteína llamada
transcobalamina que la lleva a la médula ósea para intervenir en la formación
de los hematíes. La porción que no se usa se almacena en el hígado.
La falta de vitamina B12 puede ser debida a un
déficit de la misma en la dieta, por una ingesta pobre en productos de origen
animal, o a un defecto en su absorción intestinal por falta de factor
intrínseco, alteraciones en sus receptores, alteraciones de la mucosa
intestinal, enfermedades pancreáticas o infecciones intestinales. También puede
ser causada por mal uso de la vitamina B12 o bien por un aumento de las
necesidades de la misma, como en el embarazo, por tumores, hipertiroidismo o
ciertos fármacos.
De todos modos, la causa más habitual de falta de
vitamina B12 es la gastritis crónica atrófica, es decir, una atrofia de larga
evolución de la mucosa del estómago que impide que se absorba la vitamina. La
atrofia puede ser debida a una enfermedad autoinmune denominada anemia
perniciosa, en la cual el organismo crea anticuerpos contra las células de la
mucosa, cosa que impide la formación de factor intrínseco y por lo tanto de la
absorción de vitamina B12. Además, al existir una atrofia de la mucosa, se
producen menos jugos gástricos, cosa que también dificulta la absorción de
hierro, hecho que puede agravar la anemia.
Diagnóstico
En la analítica de la anemia megaloblástica se
apreciará un descenso de los niveles de hemoglobina normales.
Característicamente los hematíes de la anemia megaloblástica se hacen más
grandes y contienen mayor cantidad de hemoglobina; esto se valora con unos
parámetros concretos, que son el volumen corpuscular medio (VCM), que valora el
tamaño medio de los hematíes, y la hemoglobina corpuscular media (HCM), que
mide la cantidad media de hemoglobina por hematíe. Dado que existe un aumento
tanto del VCM como de la HCM, se clasifica la anemia megaloblástica como una
anemia macrocítica (células más grandes) e hipercroma (mayor cantidad de
hemoglobina).
También se valorarán los reticulocitos en sangre,
es decir, las formas jóvenes de hematíes existentes en sangre, que en este caso
no estarán elevados. También es característico encontrar un tipo de neutrófilos
y un aumento de una enzima llamada lactatodeshidrogenasa (LDH) a causa de la
destrucción que se produce de hematíes.
 |
| Recuperado de: http://www.clinicadam.com/imagenes-de-salud/1214.html |
LEUCEMIAS
LEUCEMIA MIELOIDE AGUDA:
Definición: LMA sin evidencia de
diferenciación mieloide por morfología o citoquímica. La naturaleza mieloide de
los blastos se demuestra mediante marcadores inmunológicos y/o estudios
ultraestructurales incluyendo citoquímica ultraestructural.
Morfología y Citoquímica: Blastos de mediano
tamaño, cromatina dispersa, núcleo redondo o ligeramente indentado con uno o
dos nucléolos. Citoplasma agranular con basofilia variable. Más raramente
presentan morfología similar a un linfoblasto. Mieloperoxidasa (MPO), Sudán
negro B (SNB) y esterasa cloroacetato negativas.
Inmunofenotipo: CD13, 33 y/o
anti-MPO +, CD3,22,79ª - ; CD34, HLA-DR + ; CD11b,14 +/-. TdT + (33%).
Genética:No anomalías
cromosómicas.Las anormalidades más frecuente son cariotipo complejo, trisomía 8
y 4. No reordenamiento de IgH ni TCR..
Halazgos clínicos: la que peor
pronóstico tiene de todas las LMA.
Equivalente no neoplásica: Célula
hematopoyética precursora en la fase más temprana de diferenciación y
maduración mieloide.
 |
| Recuperado de http://anabible.webethan.org/spip.php?article3441 |
Definición: Caracterizada por un
alto porcentaje de blastos en médula ósea sin evidencia de maduración. En el
tipo I y II constituyen más del 90% de las células no eritroides.
Morfología y Citoquímica: La mayoría presentan
mieloblasos típicos con granulación azurofílica y/o presencia de bastones de
Auer y MPO o SNB positivas. En menos casos los blastos parecen linfoblastos sin
gránulos azurófilos; la MPO y el SNB están presentes al menos en más del 3% de
las células.
Inmunofenotipo : Al menos dos
marcadores mielomonocíticos (CD13, 33, w65 y/o 1 17) y otros marcadores sin
especificidad mieloide (HLA-DR, CD34, CD7,CD4, CD15,CD11b, CD11c).
Genética: No anormalidades
cromosómicas específicas conocidas.
Hallazgos Clínicos: Generalmente se
presentan con pancitopenia.
 |
| Recuperado de http://www.medlabmaven.com/2013_04_01_archive.html |
Subtipos: LMA-M2 con
t(8;21); LMA-M2-baso.
Definición:La LMA-M2 se define
por la presencia de ³ 30% de blastos en sangre con ³ 10% de
neutrófilos maduros. Los monocitos son £ 20% de las células de la
médula ósea. En la médula se observan promielocitos, mielocitos y neutrófilos
maduros con un grado variable de displasia. La LMA-M2 es el tipo morfológico
que se asocia predominantemente con la t(8;21) y es más frecuente en niños que
en adultos; se han descrito algunos casos también con t(8;21) en la LMA-M1 o la
LMA-M4.
Genética: Translocación t
(8;21) (q22;q22) afectando a los genes LMA1 y ETO. Algunos casos de LMA-M2
tienen reordenamiento para LMA1 y ETO y son citogéneticamente negativos para la
translocación t(8;21).
Las delecciones y translocaciones afectando al
cromosoma 12 p, banda 11-13, tales como del(12)(p11p13) se asocian a LMA-M2 o
M4 con basofilia. t(6;9) (p21--22;q34), se asocia a la LMA-M2 o M4 (o síndrome
mielodisplásico SMD) con aumento de basófilos en médula ósea. La translocación
t(6;9) (q23;q24) da lugar a la formación de un gen quimérico de fusión: DEK y
CAN.
Inmunofenotipo: CD19 +, TdT +.
Hallazgos clínicos: Los pacientes con
LMA-M2 con t(8;21) tienen porcentajes de remisión completa elevadas
especialmente con dosis altas de citostáticos. Puede presentarse como sarcoma
granulocítico extramedular.
 |
| Recuperado de http://www.monografias.com/trabajos89/monografia-enfermedades-leucemia/monografia-enfermedades-leucemia.shtml |
Definición: LMA con proliferación de blastos y
promielocitos anormales.
Morfología y Citoquímica: Promielocitos hipergranulares con núcleo
reniforme o bilobado y citoplasma denso con gránulos azurófilos. El porcentaje
de blastos puede ser inferior al 30%. MPO intensamente positiva. Dos subtipos:
- LMA-M3
hipergranular
- LMA-M3 microgranular
Caracterizada por un contaje leucocitario elevado.
Los blastos y los promielocitos anormales tiene características morfológicas
distintivas como gránulos finos y núcleo bilobulado y se asemejan a monocitos.
Generalmente se identifican células "faggot" (que contienen haces de
bastones de Auer).
Inmunofenotipo: LMA M3 con t(15;17):
marcadores mieloides +, HLA-DR - (75%)
Genética: t(11;17) (q23;q21) la más frecuente; otros
casos muestran t(5;17)(q32;q12) con reordenamiento del gen RARA.
Hallazgos Clínicos: Frecuentemente se asocia a CID.
 |
| Recuperado de http://www.lookfordiagnosis.com/mesh_info.php?term=leukemia%2C+myeloid%2C+accelerated+phase&lang=1 |
Subtipos: LMA-M4Eo
Definición: Proliferación de
precursores de neutrófilos y monocitos. El diagnóstico y la diferenciación de
la LMA-M4 de la LMA-M2 y la LMA-M5b requiere la evaluación de sangre periférica
y de aspirados medulares. La médula ósea contiene ³ 30% de blastos; los
monocitos y precursores de los monocitos son ³ 20%.. Un elevado
porcentaje de monocitos pueden estar presentes en sangre periférica >5x109/L.
Morfología y Citoquímica: No siempre pueden distinguirse los monocitos y
promonocitos. El criterio citoquímico para determinar la diferenciación
monocitoide es la presencia de actividad esterasa no específica.
Inmunofenotipo: No hay marcadores
específicos para diferenciación de línea monocítica. La combinación de los
siguientes marcadores es un buen indicador: CD14,15,4, 11b, 11c, 13 y 33 +.
LMA-M4EO: inv/de16(q22)
Entidad diferenciada caracterizada por un componente eosinofílico
anormal en la m.o. y anormalidades cromosómicas específicas.
Esta misma alteración cromosómica puede encontrarse
ocasionalmente en otras leucemias mieloides como la LMA-M2 o M4 sin
eosinofilia, M5, síndrome mielodisplásico (SMD) y la crisis blástica de la
leucemia mielógena crónica.
Genética: Clonación del 16p y
16q; genes alterados CBFb y MYH 11.
Hallazgos Clínicos: Los pacientes con LMA-M4 con inv(16) tienen
una alto porcentaje de remisión completa particularmente con el uso de dosis
altas de arabinosido de citosina.
 |
| Recuperado de http://www.lookfordiagnosis.com/mesh_info.php?term=leucemia+mieloide&lang=2 |
Definición: 80% o más de las
células son de la línea monocítica, incluyendo monoblastos, promonocitos,
monocitos. Puede haber un componente menor de neutrófilos. La LMA se divide en
2 tipos: M5A (más del 80% de monocitos o monoblastos en
la m.o.) y M5B (menos del 80% son monoblastos y la
célula que predomina son los promonocitos).
Morfología y
Citoquímica: Monoblastos: células grandes,
citoplasma abundante basofílico, con algunos gránulos azurofílicos y vacuolas.
Núcleo redondo, cromatina laxa y uno o más nucléolos grandes. Promonocitos: Núcleo más irregular y
ligeramente convoluto; citoplasma generalmente menos basofílico; ocasionales
gránulos grandes azurófilos. Los bastones de Auer son raros en la LMA5 y cuando
se ven están en células que se identifican como mieloblastos. En la mayoría de
los casos los monoblastos y promonocitos muestran actividad esterasa no
específica intensamente positiva. En el 10-20% de los casos, más frecuentemente
en la M5A, la reacción no específica es negativa o muy débilmente positiva.
Inmunofenotipo: No hay marcadores específicos para la línea
monocítica. Sin embargo son indicadores de diferenciación monocítica la
expresión de: CD14,15,4,11b, 11c, y CD68, junto a CD13 y CD33.
Genética: Delecciones y
translocaciones del cromosoma 11 band q23. Casos aislados de M5B, M4, M2, M1 y
LLA pueden mostrar la misma anormalidad.
Translocación t(8:16)(p11;pl3) en la mayoría de los
casos, junto a características de hemofagocitosis por las células leucémicas,
particularmente eritrofagocitosis.
Hallazgos Clínicos: Se presenta con
manifestaciones cutáneas, infiltración visceral y afectación del SNC. La
LMA-M5A es particularmente frecuente en niños y neonatos y se asocia con
frecuencia a reordenamiento del cromosoma 11q23. Puede presentarse como un
sarcoma monoblástico extramedular.
 |
| Recuperado de http://tefacilitamoslainformacion.blogspot.com/ |
LMA M6a: presencia
de más del 50% de las células en m.o. son precursores eritroides
y ³ 30% de mieloblastos en la población no eritroide.
LMA M6b: Proliferación
neoplásica de células inmaduras casi exclusivamente de la línea eritroide (más
del 80%) sin evidencia de componente mieloblástico significativo.
Morfología y Citoquímica:
LMA-M6a: Precursores
eritroides en todos los estadios frecuentemente con tendencia a la inmadurez.
Blastos similares a mieloblastos. PAS + en precursores eritroides.
LMA-M6b: Con
o sin características de diferenciación eritroide. Diseritropoyesis marcada.
Normoblastemia.
Inmunofenotipo: LMA- M6a: CD13
+, CD33 +, anti-MPO +, CD34 +/-, HLA-DR II +/-. LMA- M6b: Los casos
más diferenciados pueden mostrar glicoforina A y hemoglobina A y negatividad
para marcadores mieloides. Los blastos a menudo son negativos para HLA-DR II y
CD34. Las formas más inmaduras son negativas para anti-glicoforina A o
débilmente positiva en una minoría de blastos.
Genética: No anomalía
cromosómica descrita.
Hallazgos Clínicos: Anemia intensa y
normoblastemia. La LMA-M6a puede ser primaria o secundaria a un sd.
mielodisplásico.
Equivalente no
Neoplásico: LMA-M6a: Célula precursora
multipotencial (stem cell) con amplio potencial mieloide. LMA-M6b: Célula
primitiva (BFU-E/CFU-E) con cierto grado de diferenciación hacia línea
eritroide.
 |
| Recuperado de http://apuntesdemedicina.awardspace.com/Leucemias_clasificacion.htm |
Definición: 30% de blastos
de la línea megacariocítica en médula ósea y/o sangre.
Morfología y
Citoquímica: Sangre periférica: blastos de
mediano a gran tamaño, con núcleo redondo, o indentado con 1-3 nucléolos.
Citoplasma basofílico, a menudo agranular con pseudópodos prominentes. Médula
ósea: variable desde casos con una población uniforme de blastos pobremente
diferenciados a una mezcla de blastos y megacariocitos displásicos madurando.
Fibrosis retículínica variable. SNB y MPO son negativas. PAS, fosfatasa ácida y
esterasa no específica positivas.
Inmunofenotipo: Marcadores linfoides, TdT, CD34, HLA-IIDr
negativos. Expresión de uno o más glicoproteínas plaquetarias: CD41, CD42, y/o
CD61. Marcadores mieloides CD13 y 33 + (33%); MPO -. En m.o. blastos y
megacariocitos son factor VIII +.
Genética: Varias
anormalidades genéticas descritas en adultos, tales como inv(3)(q21;q26). En
niños, particularmente en menores de 1 año, puede haber t(1;22)(p13;q13) con rasgos clínicos distintivos.
Hallazgos Clínicos: Ocurre en adultos
y niños. Generalmente se presenta con trombocitopenia y en algunos casos
trombocitosis. La hepatoesplenomegalia es infrecuente excepto en niños con
t(1;22). Pronóstico pobre, particularmente en estos últimos casos.
Equivalente no Neoplásico: Célula precursora hematopoyética relacionada
con la línea megacariocítica y/o capaz de diferenciación hacia esa línea.
 |
| Recuperado de http://dc378.4shared.com/doc/9ktf5rPs/preview.html |
LEUCEMIA LINFOIDE AGUDA:
·
LEUCEMIA LINFOIDE AGUDA (L1) - Alrededor 25 a los 30% de casos
adultos y los 85% de casos de la niñez de TODOS están de este subtipo.
Dimensión de una variable nuclear regular
Cromatina homogénea
Nucléolo pequeño o ausente
Forma nuclear: Regular con
hendiduras o muescas ocasionales.
Citoplasma escaso y
ligeramente basófilo, con vacuolas citoplasmáticas.
Inmunofenotipo: Tdt +, CD19, 22, 20, 79, 45 y 10 +/-. CD22 c es
específica de línea.
Genética: Diversas anormalidades citogenéticas asociadas de significado
pronóstico. En niños más del 50% de los casos tienen hiperdiploidia y t(12;21)
con buen pronóstico y supervivencia del 85-90%. Otros casos se asocian a
translocaciones del gen MLL
en el 11q23, indicador de mal pronóstico independientemente de la edad. En los
adultos: t(9;22) en el 25% de los casos con mal pronóstico; las translocaciones
11q23 son más frecuentes; la hiperdiploidia asociada a buen pronóstico con
51-65 cromosomas y t(12;21) son infrecuentes.
 |
| Recuperado de http://dc349.4shared.com/doc/z1jYt8Wc/preview.html |
·
LEUCEMIA LINFOIDE AGUDA (L2): Los Alrededor 70% de casos adultos y
los 14% de casos de la niñez son de este tipo.
Tamaño: Las células son
grandes y o las dimensiones de una variable variadas con: -
Cromatina: heterogénea, variable
Nucléolo: más de uno, grandes
Cantidad de citoplasma:
Moderadamente abundante
Basofilia citoplasmática:
Variable, intenso.
Vacuolización citoplasmática:
Variable.
Inmunofenotipo: CD1a, CD2,3,5 y 7 +variable. CD3 es el marcador T más
temprano y específico.
Genética: Anormalidades genéticas sin significado pronóstico: (33%)
translocaciones del TCR con genes para transcripción de diversos factores (MYC,
TAL1, RBTN2, RBTN2, HOX11 y LCK); (30%) delección 9p-.
Hallazgos Clínicos: 15% de las LLA en
niños; más frecuente en adolescentes que en niños. 25% de las LLA del adulto.
Mal pronóstico independientemente de los hallazgos citogenéticos. Se presentan
como masa mediastínica o de otros tejidos y leucocitosis elevada en sangre.
Equivalente no Neoplásica: Células precursoras T o células T en
diferentes estadios madurativos
 |
| Recuperado de http://dc349.4shared.com/doc/z1jYt8Wc/preview.html |
·
LEUCEMIA LINFOIDE AGUDA (L3) Esto es un subtipo más raro con los
solamente casos de 1 a del 2%.
Tamaño: Grande, homogéneo.
Cromatina nuclear: Finamente punteada y homogénea.
Forma nuclear: Regular,
redondea.
Nucléolos: Prominentes
Cantidad de citoplasma: Moderadamente
abundante
Basofilia citoplasmática:
muy intensa.
Vacuolización: Prominente.
En este tipo las
células son grandes y uniformes con las vacuolas (burbuja como características)
en el citoplasma que cubre el núcleo.
Inmunofenotipo:TdT -, Igs con restricción de cadenas. Generalmente
CD19,20,22 y 79 +.
Genética: Anormalidades citogenéticas idénticas a las del linfoma de Burkitt.
Equivalente no Neoplásico: Célula precursora temprana B con inmunoglobulina
de superficie.
Hallazgos Clínicos: Representa la forma leucémica del linfoma de Burkitt. Se
presenta con tumor grande y LDH elevada. Con tratamiento agresivo los pacientes
tienen buen pronóstico, supervivencia del 80-90%.
 |
| Recuperado de http://www.lookfordiagnosis.com/mesh_info.php?term=leucemia+promieloc%C3%ADtica+aguda&lang=2 |
LEUCEMIAS
CRONICAS
Definición:Síndrome mielodisplásico con monocitosis persistente mayor
de 1 x 109 /L.
Morfología: Los hallazgos en sangre varían desde únicamente monocitosis
a marcados cambios displásicos en los neutrófilos, plaquetas y hematíes.
Blastos en sangre < 5%. No hay bastones de Auer. La médula ósea muestra un
número variable de monocitos. Los mieloblastos pueden estar aumentados pero el
número de blastos y promonocitos no excede el 20%.
Citoquímica: Los monocitos son esterasa no específica positivos; MPO
dispersa positiva y gránulos SNB positivos.
Genética: No anormalidades citogenéticas específicas identificadas.
Algunas anormalidades cromosómicas inespecíficas como trisomía 8, monosomía 7 y
20q-.
Hallazgos Clínicos: Afecta a viejos. Puede haber esplenomegalia. El 50% tienen
hipergammaglobulinemia. Supervivencia media de 11 a 36 meses. Progresión a
leucemia aguda en 14-24% de los casos.
 |
| Recuperado de http://www.lookfordiagnosis.com/mesh_info.php?term=Leucemia+Mielomonoc%C3%ADtica+Cr%C3%B3nica&lang=2 |
·
LINFOMA DE
LINFOCITOS PEQUEÑOS B (LLP-B)
Morfología:
Linfocitos B pequeños monocromos con una
mezcla de prolinfocitos y parainmunoblastos y formación de pseudofolículos o
centros de proliferación. Mitosis escasas. Ocasionalmente los núcleos presentan
moderada irregularidad que plantea diagnostico diferencial con linfomas de
células del manto. El término de LLP-B se utiliza solamente en los casos en que
la morfología y el inmunofenotipo son de LLC-B pero no hay leucemia.
Inmunofenotipo:SIg, CD20, 19,79a, 22+; CD5,CD23, CD43+. CD10-.CD23 es útil
para distinguirlo del linfoma de células del manto.
Genética: Trisomía 12 (33%), alteraciones en 13q (25%),
reordenamiento de IgH y IgL
Hallazgos
Clínicos: Adultos con curso indolente.
Generalmente afectación de médula ósea y sangre periférica en el momento del
diagnóstico. Generalmente no curable con la terapia. Puede trasformarse a un
linfoma de células grandes (Síndrome de Richter).
Equivalente
no Neoplásico: Célula B periférica
recirculante CD5+ y CD23+.
 |
| Recuperado de http://zl.elsevier.es/es/revista/gastroenterologia-hepatologia-14/linfoma-manto-colon-13083907-cartas-al-director-2006 |
DESORDENES
LINFORPOLIFERATIVOS:
·
(MACROGLOBULINEMIA
DE WALDENSTROM +/-)
Morfología:
Neoplasia de linfocitos pequeños B,
linfocitos plasmacitoides y células plasmáticas. Se excluyen las variantes
plasmacitoides/cíticas de otros linfomas.
Inmunofenotipo: SIgM +, CIg +, CD5 -, CD10 -, CD19,20,22,79a +, CD23-/+,
CD43+/-. La ausencia de CD5 y la presencia de CIg son útiles para el
diagnóstico diferencial con la LLC-B.
Genética:
No anomalías específicas conocidas.
Reordenamiento de genes de IgH y IgL.
Hallazgos
Clínicos: Adultos con afectación de médula
ósea, ganglios linfáticos y bazo. La mayoría de los pacientes tienen una
paraproteína sérica IgM monoclonal con síntomas de hiperviscosidad, fenómenos
autoinmunes o crioglobulinemia.. Curso indolente generalmente no
curable.
Equivalente
no neoplásico
Linfocito
B CD5- periférico, en diferenciación a célula plasmática.
 |
| Recuperado de http://medlibes.com/entry/waldenstroms-macroglobulinemia |
Mieloma múltiple asintomático sin
lesiones osteolíticas. En algunos pacientes el mieloma sintomático no se
desarrolla hasta después de muchos años. No se trata.
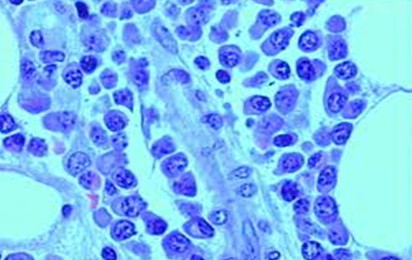 |
| Recuperado de http://www.portalesmedicos.com/publicaciones/articles/4753/1/Mieloma-multiple.html |
Esencialmente igual al quiescente
excepto que presenta tres o menos lesiones líticas. Tampoco se trata.
 |
| Recuperado de http://fundacionjosepcarreras.blogspot.com/2011_09_01_archive.html |
Representa el 2% de los MM. Células
plasmáticas circulantes >20% de los leucocitos de la sangre. Pueden ser
casos primarios o complicación terminal del MM. En el inmunofenotipo no
expresan CD56.
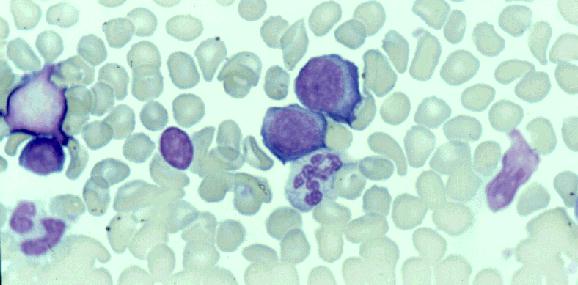 |
| Recuperado de http://www.lookfordiagnosis.com/mesh_info.php?term=Leucemia+de+C%C3%A9lulas+Plasm%C3%A1ticas&lang=2 |
Síndrome constituido por
Polineuropatía, Organomegalia (hepatoesplenomegalia), Endocrinopatía,
gammapatía Monoclonal y lesiones en la piel ("Skin"). El síntoma
predominante es una polineuropatía desmielinizante crónica y presentan única o
múltiples lesiones osteoscleróticas.
 |
| Recuperado de http://zl.elsevier.es/es/revista/radiologia-119/sindrome-poems-proposito-un-caso-13140139-comunicaciones-breves-2009 |
Representa el 1% de los MM. Sintetiza
pero no secreta Ig (ausencia de Ig monoclonal en suero u orina). Clínica
idéntica a la del MM.
 |
| Recuperado de http://www.bindingsite.es/identificacion-rapida-de-pacientes-con-riesgo-de-dano-renal |
Morfología:> 10% de células plasmáticas maduras o inmaduras
(plasmablastos) en médula ósea (m.o.). El grado de infiltración tiene valor
pronóstico; tres estadios: I: <20%; II 20-50%; III >50% de células
plasmáticas en m.o.
Inmunofenotipo:SIg - , CIg +, CD 38 +, CD5 -, CD10 -, Pan B -, CD45-, CD56
+,. CD30 +, IL-1 +.
Genética:No anomalías específicas conocidas.
Hallazgos Clínicos:Afecta a adultos. Lesiones líticas múltiples, fracturas
patológicas, hipercalcemia y anemia. Proteína M en suero u orina en 99% de los
casos.
Equivalente no
Neoplásico:Células plasmáticas maduras.
 |
| Recuperado de http://www.medkohealth.com/condiciones/mieloma-multiple |
Morfología: Células plasmáticas maduras constituyendo menos del 10% de
las células nucleadas de la médula ósea.
Inmunofenitpo:CIg +, CD38 +, CD56, IL-1 -, Pan B -, CD45 -.
Genética:Ninguna anomalía específica conocida.
Hallazgos
Clínicos:Asintomáticos. Presencia de
gammapatía monoclonal sin evidencia de mieloma múltiple, macroglobulinemia de
Waldenström, amiloidosis primaria u otros procesos relacionados. Un 25%
evolucionará a alguna de estas enfermedades después de un intervalo medio de 10
años.
Equivalente
no Neoplásicos:
Célula
plasmática madura.
 |
| Recuperado de http://www.edicionesmedicas.com.ar/Actualidad/Ultimas_noticias/Factores_que_inciden_en_el_mieloma_multiple |
TRANSTORNOS
MIELOPROLIFERATIVOS
·
Trombocitemia esencial o primaria:
• La
TE es un SMPc caracterizado por una proliferación excesiva de megacariocitos y una trombocitosis importante
con un curso relativamente favorable. La
mayoría de los pacientes se presentan asintomáticos, pero los síntomas
fundamentales son producto de fenómenos tromboembólicos y microvasculares. El riesgo de trombosis puede
llegar hasta 50 % en pacientes con
historia previa de trombosis y es mucho menor cuando está ausente este antecedente.La expectativa de
vida se señala igual que la población general, pero en pacientes jóvenes esta se ve disminuida, así
como la calidad de vida. La progresión a una leucemia aguda es una posibilidad
con el transcurso de los años y puede
presentarse en su evolución natural o relacionada con el tipo de tratamiento
utilizado.La trombocitosis en general es producida por tres causas
fundamentales: pueden ser reactivas o secundarias, puede ser una trombocitosis
familiar o pueden ser clonales. La TE se diagnóstica por la exclusión de otras
causas de trombocitosis.
Causas de trombocitosis
•
Trombocitosis reactivas
—
Déficit de Fe
—
Infecciones o inflamaciones crónicas (conectivopatías, arteritis temporal,
tuberculosis, bronconuemopatías crónicas, vasculitis)
—
Infecciones e inflamaciones agudas.
—
Hemorragias agudas
—
Anemias hemolíticas
—
Respuesta al ejercicio
—
Post-esplenectomía
— Daño
tisular (cirugía, infarto agudo miocárdico, pancreatitis, traumas)
—
Neoplasias
—
Recuperación de una trombocitopenia (rebote)
—
Respuesta a drogas (vincristina, ácido retinoico, epinefrina, factor de crecimiento)
•
Trombocitosis familiar
•
Trombocitosis clonales
—
Trombocitemia esencial
—
Otros SMPc (LMC, PV, MP)
Aspectos clínicos y biológicos de la TE
• En
la mayoría de los pacientes se detecta la trombocitosis de manera fortuita, en exámenes de rutina, estando
estos asintomáticos.
• En
los que presentan síntomas predominan las manifestaciones trombohemorrágicas y alteraciones de la
microcirculación. Las primeras son la
mayor causa de morbimortalidad, conjuntamente con la transformación maligna en la evolución natural de la
enfermedad o por causa del tipo de
tratamiento realizado.
• La
TE es la causa mas frecuente de trombocitosis, con una incidencia aproximada de 2,5/100 000 habitantes. La
media de edad al diagnóstico es de 60
años y hay un ligero predominio del sexo femenino. Con la introducción de los contadores automatizados
de plaquetas han aumentado los casos de
pacientes jóvenes asintomáticos. Han sido descritos casos excepcionales en la
infancia.
La trombosis es
reportada en algunas series hasta en 50 % de los casos en algún momento de la
evolución de la enfermedad, con más frecuencia en pacientes mayores de 60 años
con algún factor de riesgo trombogénico asociado, como: HTA,
hipercolesterolemia, diabetes mellitus o el hábito de fumar. Igualmente en
aquellos pacientes que además de la TE portan una trombofilia o un trastorno de
hipercoagulabilidad adquirido. Las trombosis pueden ser arteriales o venosas,
siendo las trombosis venosas profundas de MI de las más comunes.
• Los
fenómenos trombóticos pueden ser mayores como son: accidentes vasculares
encefálicos, infarto del miocardio, angina de pecho, trombosis de la arteria
central de la retina, trombosis arteriales periféricas, trombosis venosas
profundas y tromboembolismo pulmonar.La trombosis es reportada en algunas
series hasta en 50 % de los casos en algún momento de la evolución de la
enfermedad, con más frecuencia en pacientes mayores de 60 años con algún factor
de riesgo trombogénico asociado, como: HTA, hipercolesterolemia, diabetes
mellitus o el hábito de fumar. Igualmente en aquellos pacientes que además de
la TE portan una trombofilia o un trastorno de hipercoagulabilidad adquirido.
Las trombosis pueden ser arteriales o venosas, siendo las trombosis venosas
profundas de MI de las más comunes.
Diagnostico:
—
Cuantificación de fibrinógeno
—
Proteína C reactiva
— Ferritina
sérica
—
Radiografía de tórax.
—
Ultrasonido abdominal.
—
Serologias: VDRL, HIV, VHB, HBC.
—
Electrocardiograma.
—
Cituria.
—
Electroforesis de proteínas.
Tratamiento: Para tomar la decisión terapéutica debe considerarse la
edad del paciente, el riesgo de trombosis relacionado con la TE y el riesgo de
trombosis no relacionado con la TE.Existe una clasificación por el riesgo que
puede tener el paciente de padecer una trombosis mayor o sangramiento
importante:
Medicamentos
•
Hidroxiurea (HU): 15-30 mg/kg: respuesta de 85 %. Reduce el número de plaquetas
en 4 s. Después se valora una dosis de mantenimiento. Posibilidad de
transformación maligna, asociación con el síndrome 17p-. No se han visto
malignidades en miles de pacientes tratados por enfermedades no malignas (HB SS
y cianosis congénita), pero estas no son enfermedades clonales.
•
Interferón alfa recombinante (IFaR): dosis más usada: 21 millones deU (MU)
semanales. Se puede utilizar una dosis de 3 MU/m2/día.
•
Anagrelide: compuesto imidazoquinolina oral. Actúa en la fase post mitótica del
megacariocito inhibiendo su maduración. Dosis diaria de 1-4 mg. Dosis inicial
de 0,5 mg cuatro veces al día, se sube semanalmente 0,5 mg hasta lograr el
efecto deseado. Respuesta de 93 %. Efectos secundarios fundamentales (cefalea,
taquicardia con palpitaciones, retención hídrica,intolerancia
gastrointestinal). Debe usarse con precaución en gestantes porque atraviesa la
barrera placentaria.
•
Pipobroman: tiene una estructura química similar a los agentes alquilantes.
Usado fundamentalmente en Europa en la TE y en la PV. Dosis diaria inicial de
0,8-1 mg/kg/día. Después de una inducción media de alrededor de 60 días se deja
un mantenimiento de 0,3-0,6 mg/kg/día.
Respuesta
de 95 %.
•
Busulfán: a las dosis empleadas en la LMC. Transformación a leucemia aguda.
•
Fósforo radiactivo: 2,3 mc/mq. Desarrollo de segundas neoplasias.
Melfalán:
a las dosis habituales. Relacionado con transformación a leucemia aguda.
•
Aféresis terapéutica de plaquetas.
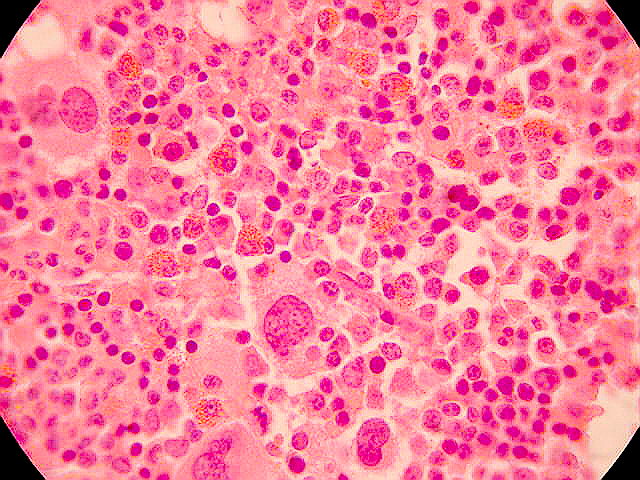 |
| Recuperado de http://www.conganat.org/7congreso/trabajo.asp?id_trabajo=336&tipo=2 |
·
Policitemia vera
•
La PV es una enfermedad de la célula madre hematopoyética (stem cell), de
inicio insidioso, curso crónico y causa desconocida. Esta caracterizada por
proliferación excesiva y sostenida en la médula ósea de células eritroides, granulocíticas y
megacariociticas. Por definición, la proliferación eritroide es dominante, no
influida por su factor de crecimiento específico, y se expresa por un aumento
absoluto del volumen eritrocitario. Tiene una incidencia de 2,3/ 100 000, con
una edad media al diagnóstico de 60 años y un ligero predominio del sexo
masculino. La expectativa de vida puede exceder los 10 años y la global es
ligeramente menor que en los pacientes normales tomados como control,
relacionado con las muertes prematuras por cuadros trombohemorrágicos y su transformación
en LMA, fundamentalmente.Etiopatogenia
• La PV es un SMPc de etiología desconocida que se origina de un progenitor hematopoyético pluripotencial. Está caracterizado por una producción anormal de eritrocitos, leucocitos y plaquetas en ausencia de un estímulo fisiológico reconocible.
Cuadro clínicoAunque no es una enfermedad eminentemente mortal, sí afecta la calidad de vida de los pacientes que la padecen. Las alteraciones clínicas y de laboratorio que pueden verse son:
• Leucocitosis persistente.
• Trombocitosis persistente.
• Microcitosis por déficit de Fe.
• Esplenomegalia.
• Prurito generalizado (posterior al baño).Manual de Prácticas Médicas - Hospital Hermanos Ameijeiras 20
• Trombosis inusuales.
• Eritromelalgia.
Las complicaciones más graves en la PV incluyen la trombosis y las hemorragias, principales causas de morbimortalidad. Las trombosis se producen tanto en la microcirculación, como en los grandes vasos. Hasta 20 % de los pacientes puede tener algún episodio trombótico:
• AVE
• ICT
• Oclusión de la vena o arteria de la retina,
• Isquemia coronaria,
• TEP,
• Trombosis de la vena portal o hepática,
• Trombosis venosas profundas
• Isquemias digitales
Su incidencia depende de la edad del paciente, la historia anterior de trombosis y la presencia de factores de riesgo aterosclerótico. Las complicaciones hemorrágicas son menos frecuentes y su aparición está casi siempre relacionada con el uso de AINES y el ASA.
La transformación de la enfermedad en una LMA es una complicación posible que puede presentarse, llevando al paciente a una evolución fatal.
Diagnóstico: Debe distinguirse entre una eritrocitosis secundaria, dependiente de la eritropoyetina (Epo) y la que no depende de esta y que provoca un aumento del volumen globular eritroide absoluto.
•No existen marcadores histológicos ni clonales disponibles que identifiquen inequívocamente a la PV.
• Las anormalidades citogenéticas están presentes en menos de 30 % de los pacientes al diagnóstico y tampoco son específicos.
• Además la BMO y su histología pueden ser normal o indistinguibles de la MPy la TE.
 |
| Recuperado de: http://www.lookfordiagnosis.com/mesh_info.php?term=policitemia+vera&lang=2 |
·
Mielofibrosis primaria
•
La MP ha recibido diferentes nombres y los más comunes son metaplasia mieloide
agnogénica, osteosclerosis, mielosis crónica no leucémica, etc. En la MP hay
una expansión clonal de la célula madre hematopoyética que seacompaña de una
proliferación reactiva no-clonal de los fibroblastos y fibrosis de la médula
ósea. A medida que la médula ósea se hace fibrótica la hematopoyesis no se
puede mantener y ocurre una hematopoyesis extramedular (metaplasia mieloide) en
el hígado y el bazo. Es el SMPc de más baja prevalencia y de más difícil manejoCaracterísticas clínicas
• En ocasiones el paciente acude al médico por notarse el mismo una masa en el abdomen o por síntomas atribuibles a la esplenomegalia como molestias o dolor. Pueden ocurrir infartos esplénicos, periesplenitis o hematomas subcapsulares que provocan dolor intenso en el cuadrante superior izquierdo del abdomen y/o en el hombro de ese lado. Otros síntomas relacionados con la esplenomegalia son las diarreas por compresión del colon, la sensación precoz de llenura con los alimentos por compresión gástrica y los edemas en miembros inferiores.• El paciente puede manifestar síntomas de anemia y síntomas generales como astenia, perdida de peso, sudores nocturnos, febrícula, que pueden ser consecuencia de un estado de hipercatabolia. Pueden observarse litiasis renal por uratos o un cuadro de gota.
• El paciente puede manifestar síntomas relacionados con la localización de la hematopoyesis extramedular: hemorragias digestivas, compresión de médula espinal, convulsiones focales, síntomas de tumor cerebral, ascitis, derrame pericárdico, derrame pleural, hemoptisis y fallo respiratorio.
• Puede presentarse un cuadro completo de hipertensión portal por un flujo esplenoportal marcadamente incrementado. Puede ocurrir una trombosis portal o hepática como complicación.
• La leucopenia se observa en menos de 25 % de los casos, mientras que la leucocitosis es frecuente, aunque es raro observar cifras mayores de 50 x109/L. Predominan los granulocitos maduros, pero se observan también formas jóvenes (mielocitos, juveniles, etc.), y un número pequeño de blastos y células de Pelger Huet.
• La trombocitosis es mas frecuente que la trombocitopenia y puede detectarse en el 35-50 % de los casos. La trombocitopenia se incrementa a medida que avanza la enfermedad. En la extensión pueden observarse megacariocitos, fragmentos de megacariocitos y plaquetas gigantes.
• En los estudios de la hemostasis es frecuente la disfunción plaquetaria y se puede detectar una CID, clínicamente silente, en 15 % de los casos
 |
| Recuperado de http://tomatetumedicina.wordpress.com/author/humbertocruz/page/12/ |
LINFOMAS
Linfoma
de Hodgkin clásico (LHc). El LHc es el tipo más
común de linfoma de Hodgkin que ocurre alrededor del 95 % del tiempo. Se
diagnostica cuando se encuentran linfocitos anormales característicos,
conocidos como células Reed-Sternberg. Como se mencionó anteriormente, el LHC
se puede dividir en cuatro subtipos diferentes:
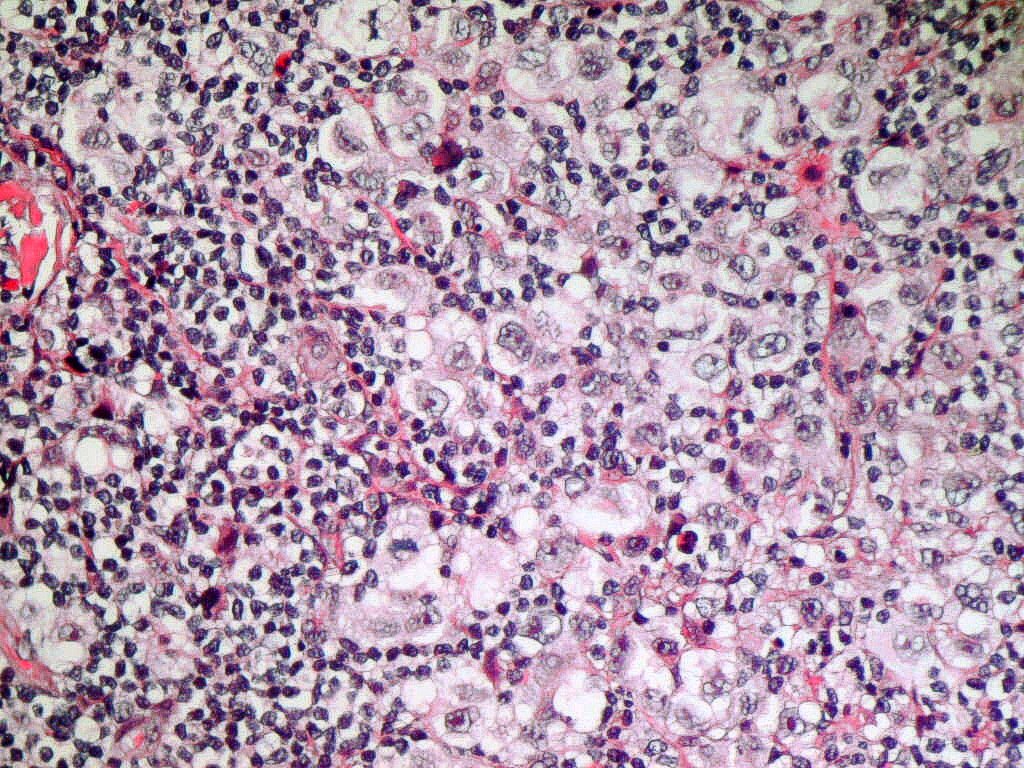 |
| Recuperado de http://www.conganat.org/seap/regional/galicia/junio2003/vigodg.htm |
 |
| Recuperado de: http://grupolinfomas.es/gallery/linfoma-de-hodgkin/ |
·
Linfoma
de Hodgkin clásico rico en linfocitos: aproximadamente
el 6% de las personas con LHc tienen este subtipo. Su aparición es más frecuente
en hombres y, generalmente, compromete otras áreas además del mediastino. El
tejido contiene muchos linfocitos normales, además de células de
Reed-Sternberg.
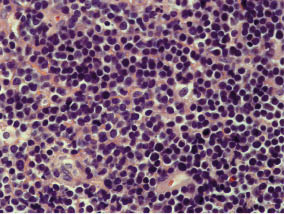 |
| Recuperado de http://www.pediatriaintegral.es/numeros-anteriores/publicacion-2012-07/linfomas-de-hodgkin-y-hodgkin/ |
·
Linfoma
de Hodgkin con celularidad mixta: este subtipo de
linfoma se presenta en adultos mayores y, frecuentemente, aparece en el
abdomen. Contiene muchos tipos diferentes de células, entre las que se incluyen
grandes cantidades de células de Reed-Sternberg.
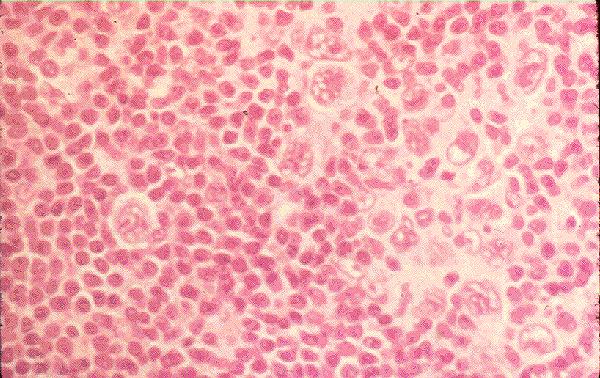 |
| Recuperado de http://www.conganat.org/linfo.tortosa/conf/cap3/tipos.htm |
·
Linfoma
de Hodgkin con agotamiento linfocítico: el linfoma de
Hodgkin con agotamiento linfocítico es el subtipo menos frecuente de LHc, y
aproximadamente el 1% de las personas con LHc tiene esta forma. Es más común en
adultos mayores; personas con el virus de inmunodeficiencia humana (VIH), el
virus que causa el síndrome de inmunodeficiencia o SIDA, y personas en países
no industrializados. El ganglio linfático contiene casi en su totalidad células
de Reed-Sternberg.
 |
| Recuperado de http://www.seom.org/en/informacion-sobre-el-cancer/info-tipos-cancer/linfomas-y-mieloma/linfoma-no-hodgkin?showall=1
ENFERMEDADES
PARASITARIAS
|
El paludismo es causado por el protozoo del género Plasmodium. Se han identificado alrededor de 150 especies pero son de relevancia las especies que se han adaptado al hospedero humano y son transmitidas principalmente por un vector anofelino:Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae y Plasmodium ovale. Plasmodium knowlesi, parásito de ciertos macacos en el sudeste de Asia, también se ha identificado como causa de enfermedad. (Cox-Singh J. 2012; Sermwittayawong et al., 2012).
Ciclo biológico:
Ciclo de vida de Plasmodium. El ciclo
de vida del parásito posee dos fases, una asexual que se da en el hospedero
humano (flechas azules) y una sexual que se da en el vector Anopheles spp.
(flechas rojas). En el humano, el ciclo empieza con la fase exoeritrocítica (A)
cuando por la picadura de mosquitos hembra del genero Anopheles, se introduce
la forma infectante del parásito (esporozoítos) al torrente sanguíneo (1).
Luego los esporozoítos migran al hígado en aproximadamente 30 minutos, donde
logran evadir la respuesta inmune de los macrófagos de Kupffer e infectan los
hepatocitos (2). Una vez dentro del hepatocito maduran, aproximadamente en 4
semanas (tiempo de incubación) a esquizontes tisulares (3). Además, en las
especies de P. vivax y P. ovale existe otra forma del parásito, el hipnozoíto
(4), donde el parásito persiste en forma latente dentro del hepatocito y es el
responsable del fenómeno de recaída meses después de la infección primaria. Con
la ruptura de los esquizontes tisulares se liberan merozoítos al torrente
sanguíneo (5), iniciando la fase eritrocítica del ciclo (B), en la cual los
parásitos se adhieren e invaden los eritro- citos (6). En los eritrocitos se
desarrolla el estado de trofozoíto que pasa por un proceso de maduración,
acumulando progresivamente hemozoína, pasando por las formas de trofozoíto
anular (7), trofozoíto maduro (8) y esquizonte (9). El esquizonte eritrocítico
acumula merozoítos y luego se rompe (10) liberando merozoítos que invadirán
otros eritroci- tos para continuar con la etapa de reproducción asexual
eritrocítica del parásito. La liberación periódica de merozoítos de los
esquizontes eritrocíticos produce los paroxismos de la malaria posteriores al
primer paroxismo. Durante la fase eritrocítica, algunos trofozoítos se
diferencian (11) a la forma sexual del parásito o gametocito masculino
(microgame- tocito) o gametocito femenino (macrogametocito) (12). Los
gametocitos circulantes son captados cuando el mosquito pica al hospedero
durante la alimentación (13) y una vez en el intestino del mosquito, los
gametocitos se fusionan para formar cigotos (14) e iniciar la parte sexual del
ciclo en el mosquito (C). El cigoto se desarrolla a ooquinetos invasores (15)
que atraviesan el intestino medio y se transforman en un ooquiste (16), que a
través de divisiones mitóticas su- cesivas, en 10 a 14 días producen miles de
esporozoítos que luego son liberados del quiste (17) y que migran a las
glándulas salivales, desde donde pueden infectar al humano para reiniciar el
ciclo (1).
Trofozoito en sangre:
 |
| Recuperado de: http://invertebradosunica.blogspot.com/2012/05/tema-03-protozoos-cilioforos-y.html |
Esquizonte:
 |
| Recuperado de: http://fcfrp.usp.br/dactb/Parasitologia/Arquivos/Genero_Plasmodium.htm |
+(Col.+Giemsa).jpg) |
| Recuperado de: http://medicinaucv.blogspot.com/2008/06/parasitologa-prctica-n-10-plasmodium.html |
Transmisión.
El paludismo se transmite por la picadura del mosquito hembra del género Anopheles, el hospedero definitivo, en el cual se lleva a cabo la fase sexuada de la reproducción del parásito.
La transfusión de sangre infectada y el empleo de agujas y jeringas contaminadas puede dar lugar a paludismo.
Es posible la infección durante una transfusión sanguínea. La transmisión puede producirse mientras circulen formas asexuales en la sangre.
En mujeres embarazadas, más vulnerables, principalmente ante infecciones por P. falciparum, puede presentarse paludismo severo, parto prematuro, aborto y transmisión congénita.
El paludismo se transmite por la picadura del mosquito hembra del género Anopheles, el hospedero definitivo, en el cual se lleva a cabo la fase sexuada de la reproducción del parásito.
La transfusión de sangre infectada y el empleo de agujas y jeringas contaminadas puede dar lugar a paludismo.
Es posible la infección durante una transfusión sanguínea. La transmisión puede producirse mientras circulen formas asexuales en la sangre.
En mujeres embarazadas, más vulnerables, principalmente ante infecciones por P. falciparum, puede presentarse paludismo severo, parto prematuro, aborto y transmisión congénita.
CHAGAS:
EtiologíaLa enfermedad de Chagas se origina a partir de la infección con el parásito protozoario Trypanosoma cruzi, miembro de la familia tripanosomatidae . La mayoría de las cepas de este parásito se pueden clasificar en 2 grupos principales, T. cruzi I y T. cruzi II, que incluso se pueden dividir en diversos linajes (p. ej., T. cruzi IIa). Los linajes tienden a asociarse a determinadas especies de huéspedes, aunque esta relación no es absoluta.
 |
| Recuperado de: http://www.facmed.unam.mx/deptos/microbiologia/parasitologia/trypanosomosis.html |
La
enfermedad de Chagas es una enfermedad transmitida por vectores, principalmente
insectos triatóminos, también llamados insectos redúvidos, “escarabajos o
insectos chupasangre”, “insectos asesinos” o “vinchucas”. Más de 130 especies
de estos insectos parecen ser capaces de transmitir T. cruzi; Triatoma,
Rhodnius y Panstrongylus se consideran las especies más importantes del género.
El parásito generalmente completa su ciclo de vida al realizar el ciclo entre
una especie de insectos y una especie de mamíferos con los que el insecto vive
en estrecha relación. Los huéspedes mamíferos incluyen especies silvestres,
animales domésticos y humanos. En su huésped mamífero, T. cruzi se puede
encontrar en la sangre como tripomastigotes (formas extracelulares no divisibles)
y en células como amastigotes (formas replicativas). Cuando un insecto succiona
la sangre de un mamífero infectado, ingiere los tripomastigotes. Luego de 2 a 4
semanas de evolución, algunos de los parásitos migran al intestino posterior,
donde se transforman en tripomastigotes metacíclicos infecciosos. El insecto
defeca luego de alimentarse y libera los
tripomastigotes en las heces. Estos parásitos pueden ingresar al
organismo del mamífero a través de las
membranas mucosas o lesiones cutáneas. Al rascarse, se pueden inocular los
tripomastigotes en la herida de la picadura o permitir que los parásitos
ingresen a través de los rasguños. Las especies de insectos triatóminos que
muestran defecación tardía luego de alimentarse tienen menos probabilidades de
transmitir la enfermedad de Chagas, que las especies que defecan sobre el
huésped. Los humanos y los animales también se pueden infectar si ingieren el
insecto o alimentos crudos que contienen heces de insectos. Existen 3 ciclos
básicos en la transmisión de T. cruzi. En el ciclo selvático (salvaje), este
organismo realiza el ciclo entre las especies silvestres y los insectos
triatóminos que habitan en ambientes selváticos. Los humanos y los animales
domésticos se infectan ocasionalmente cuando entran en contacto con estos
insectos en el hábitat natural. En ciertas circunstancias, los insectos también
pueden invadir los hogares o dependencias cuando son atraídos por la luz, el
calor o determinados olores, y pueden contaminar los alimentos. Los insectos
triatóminos silvestres también pueden ser transportados accidentalmente a los
hogares de loshumanos. El ciclo selvático es responsable de relativamente pocos
casos de enfermedad de Chagas. Es el único ciclo en EE. UU. También existe un
ciclo de transmisión doméstica en México y partes de Centroamérica y
Sudamérica. En este ciclo, algunos insectos vectores han colonizado adobes
primitivos, pastos y casas con techos de paja, lo que ocasionó la transmisión
entre humanos e insectos. Entre las especies importantes del ciclo doméstico se
incluyen Triatoma infestans, T. dimidiata y Rhodnius prolixus. Los ciclos de
transmisión entre insectos y animales domésticos (ciclos peridomésticos)
también generan la oportunidad de que el parásito infecte a humanos. T. cruzi
no se propaga entre mamíferos por contacto
casual; sin embargo, se puede transmitir directamente a través de la
sangre (p. ej., en una transfusión) y en donación de órganos. Los carnívoros
pueden adquirir este organismo cuando comen presas infectadas. La transmisión
vertical se ha registrado en perros y otros animales, tanto por vía
intrauterina como a través de la leche. La transmisión a través de la leche es
muy poco frecuente en los humanos, pero la transmisión transplacentaria puede
ocurrir en cada embarazo, y durante todas las etapas de la infección. Las
infecciones adquiridas en laboratorios generalmente ocurren cuando los
parásitos entran en contacto con las membranas mucosas o lesiones cutáneas, o
se inocula accidentalmente a través de lesiones producidas al pincharse con una
aguja, pero también es posible la transmisión por aerosoles en este entorno.
Tripomastigote:
 |
| Recuperado de: http://www.telmeds.org/atlas/parasitologia/flagelados/trypanosoma-sp/trypanosoma-cruzi/trypanosoma-cruzi-tripomastigote-en-sangre/ |
Epimastigote:
Signos clínicos |
| Recuperado de: http://pathmicro.med.sc.edu/lecture/trypanosomiasis.htm |
La fase aguda se define como el período durante el cual los parásitos se pueden encontrar fácilmente en la sangre. Muchas personas, especialmente los adultos, son asintomáticas durante esta etapa. Los síntomas de la fase aguda son sumamente variables y pueden incluir fiebre, dolor de cabeza, anorexia, malestar, mialgia, dolor en las articulaciones, debilidad, náuseas, vómitos, diarrea, hepatomegalia, esplenomegalia y linfadenopatía generalizada o localizada. En algunos casos puede ocurrir edema, ya sea generalizado o localizado, en el rostro o en las extremidades inferiores. En ocasiones, se observa un chagoma (un endurecimiento localizado indoloro) en el lugar de la piel por donde el parásito ha ingresado. Si el ingreso se produce a través de las membranas mucosas oculares, se puede producir un edema indoloro de uno u ocasionalmente ambos ojos, con frecuencia acompañado de conjuntivitis y aumento de tamaño de los ganglios linfáticos locales. Este síndrome, que también se denomina signo de Romaña, generalmente persiste durante 1 ó 2 meses. Los pacientes ocasionalmente desarrollan un sarpullido, pero este generalmente desaparece luego de varios días. En la mayoría de los casos, los signos clínicos se mejoran en semanas o meses sin tratamiento; sin embargo, algunos casos agudos pueden ser mortales.
TOXOPLASMOSIS:
El género Leishmania spp.
pertenece al subphylum Mastigophora, orden Kinetoplastida, familia
Trypanosomatidae, de acuerdo a la taxonomía tradicional. Leishmania contempla
más de 20 especies y se divide en 3 subgéneros, de acuerdo al sitio de
desarrollo del parásito en el insecto transmisor: Leishmania (Leishmania), Leishmania (Viannia)
y Leishmania (Sauroleishmania), este último de
lagartos, de acuerdo a recientes estudios de filogenia molecular. Las especies
y subespecies se agrupan dentro de complejos en constante revisión. Asimismo,
se reconocen paraleishmanias. (Schönian et al. 2010; Fraga et al. 2010).
La difícil clasificación de las múltiples especies
y subespecies de Leishmania se realiza en función de su: 1)
Biología: desarrollo en el flebótomo, crecimiento en medios de cultivo,
desarrollo en los hospederos vertebrados; 2) Bioquímica: patrones
isoenzimáticos, secuenciación de múltiples loci (multilocus enzyme typing) -
actual "estándar de oro" (Schnian et al. 2011); 3) Inmunología:
análisis parasitario con anticuerpos monoclonales; 4) Filogenia molecular,
entre otros.
Europa: La leishmaniasis cutánea, con una abundante
variedad de lesiones, se asocia a: Leishmania major, Leishmania tropica yLeishmania
aethiopica. El complejo Leishmania donovani es
causante de la enfermedad visceral.
América: La leishmaniasis cutánea localizada es causada por múltiples especies de los subgéneros Leishmania y Viannia: L. mexicana, L. braziliensis, L. panamensis, L. guyanensis, y L. peruviana. La enfermedad mucocutánea (también conocida como espundia, uta), se asocia con mayor frecuencia a L. braziliensis y L. panamensis, aunque otras especies pueden estar involucradas. L. infantum (sín. chagasi), da lugar a la enfermedad visceral.
La leishmaniasis cutánea difusa se presenta como consecuencia de factores inmunes del hospedero, asociada a ciertas especies del parásito. Involucra, hasta ahora, a L. mexicana y L. amazonensis.
América: La leishmaniasis cutánea localizada es causada por múltiples especies de los subgéneros Leishmania y Viannia: L. mexicana, L. braziliensis, L. panamensis, L. guyanensis, y L. peruviana. La enfermedad mucocutánea (también conocida como espundia, uta), se asocia con mayor frecuencia a L. braziliensis y L. panamensis, aunque otras especies pueden estar involucradas. L. infantum (sín. chagasi), da lugar a la enfermedad visceral.
La leishmaniasis cutánea difusa se presenta como consecuencia de factores inmunes del hospedero, asociada a ciertas especies del parásito. Involucra, hasta ahora, a L. mexicana y L. amazonensis.
Leishmania es un protozoo intracelular
obligado dimórfico; en los hospederos mamíferos se localiza en macrófagos y
células dendríticas (células de Langerhans en la piel).
El promastigote( metacíclico), la forma infectante, elongado, extracelular, se desarrolla y multiplica en el tracto digestivo de los insectos transmisores, pertenecientes al género Lutzomyia en América y Phlebotomus en el Viejo Mundo. Mide 10 - 20 µm, sin contar la longitud de un único flagelo, cuyo tamaño oscila entre 15 - 25 µm; presenta un gran núcleo central, ribosomas, retículo endoplásmico, aparato de Golgi, vesículas y una mitocondria. El cinetoplasto aparece como una banda granular electrodensa dentro de la extensión de la mitocondria, localizado a 1 - 2 µm del extremo anterior del parásito, de donde emerge el flagelo. El axonema que se origina en el cuerpo basal está contenido dentro del bolsillo flagelar.
El promastigote( metacíclico), la forma infectante, elongado, extracelular, se desarrolla y multiplica en el tracto digestivo de los insectos transmisores, pertenecientes al género Lutzomyia en América y Phlebotomus en el Viejo Mundo. Mide 10 - 20 µm, sin contar la longitud de un único flagelo, cuyo tamaño oscila entre 15 - 25 µm; presenta un gran núcleo central, ribosomas, retículo endoplásmico, aparato de Golgi, vesículas y una mitocondria. El cinetoplasto aparece como una banda granular electrodensa dentro de la extensión de la mitocondria, localizado a 1 - 2 µm del extremo anterior del parásito, de donde emerge el flagelo. El axonema que se origina en el cuerpo basal está contenido dentro del bolsillo flagelar.
El amastigote, la forma replicativa, redondo u oval,
intracelular, reside y se multiplica en fagolisosomas dentro de fagocitos
mononucleares de los hospederos, aunque se ha documentado la presencia de
amastigotes en neutrófilos y fibroblastos en lesiones de piel. (Laskay T, et
al. 2003). Mide 2 - 4 µm; con tinción Giemsa se aprecian un gran núcleo y
un cinetoplasto pequeño, ambos de color púrpura, y un filamento delgado que une
cinetoplasto y cuerpo basal, éste último apenas un punto visible.
El cinetoplasto es una subestructura de la gran mitocondria, con DNA único y se encuentra asociado estrechamente al bolsillo flagelar y al cuerpo basal del flagelo. La presencia del cinetoplasto da el nombre al grupo de protozoos incluidos en el orden Kinetoplastida.
Ciclo de vida:
 |
| Recuperado de: http://www.facmed.unam.mx/deptos/microbiologia/parasitologia/toxoplasmosis.html |
 |
| Recuperado de: http://web.stanford.edu/class/humbio103/ParaSites2004/Trypanosomiasis/amastigotes%20T%20cruzi.jpg |
Referencias Bibliográficas:
- Rodak, B. (2002). Hematología: Fundamentos y aplicaciones clínicas. España: Médica Panamericana.
- Maile, J.(2002). Hematología: medicina de laboratorio. España: Editorial Reverté.
- Manascero, A. (2008). Atlas de morfología celular, alteraciones y enfermedades relacionadas.